Nucleotide diversity in Central Asian Salvia L.: high-variability regions as candidate markers Нуклеотидное разнообразие центральноазиатских Salvia L.: высоковариабельные регионы как кандидатные маркеры Орталық Азиялық Salvia L. нуклеотидтік әртүрлілігі: үміткер маркерлер ретінде жоғары өзгергіштік аймақтары
AbstractАннотацияАңдатпа
Accurate species identification and reliable reconstruction of phylogenetic relationships in many plant groups remain challenging because they are often characterized by rapid evolutionary radiations, pronounced geographic structuring, and an uneven distribution of informative plastid variation across the chloroplast genome. In this context, plastome-scale analyses provide an efficient route to identify lineage-informative regions that outperform traditional single-locus markers for closely related taxa. Here we profile chloroplast nucleotide diversity (Pi) in Salvia to quantify how variation is partitioned along the chloroplast genome and to define a practical framework for marker selection targeted to Central Asian endemic lineages. Complete chloroplast genomes were aligned and analyzed using a sliding-window approach to quantify Pi across protein-coding genes (CDS) and intergenic spacers (IGS). The results show that nucleotide diversity is concentrated predominantly in non-coding regions, especially intergenic spacers, whereas most protein-coding genes remain comparatively conserved, consistent with functional constraints on plastid gene evolution. High-Pi peaks occur predominantly in non-coding spacers and SSC-associated segments, indicating that marker performance can be substantially improved by prioritizing a small panel of hotspot loci rather than relying on widely used but low-variation regions. As the most informative and consistently high-variability candidate markers for Salvia, we identified the intergenic regions ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG as well as the protein-coding loci ycf1, matK, rpl16, rpl22, ndhF. Taken together, these findings indicate that plastome nucleotide-diversity profiling offers a robust, data-driven basis for primer development, DNA barcoding, and improved phylogenetic inference in Salvia, supporting future taxonomic and biogeographic studies of Central Asian diversity.
Точная идентификация видов и надёжная реконструкция филогенетических взаимосвязей во многих группах растений остаются сложной задачей, поскольку для них характерны быстрые эволюционные радиации, выраженная географическая структурированность и неоднородное распределение информативной пластидной вариабельности по хлоропластному геному. В этом контексте пластомно-масштабные анализы представляют эффективный подход для выявления филогенетически информативных участков, превосходящих традиционные одно-локусные маркеры при исследовании близкородственных таксонов. В настоящей работе мы оценили нуклеотидное разнообразие (Pi) хлоропластного генома рода Salvia, чтобы количественно охарактеризовать распределение вариаций пластома и предложить практическую основу отбора маркеров, ориентированную на эндемичные линии Центральной Азии. Полные хлоропластные геномы были выровнены и проанализированы методом скользящего окна для оценки Pi в белок-кодирующих генах (CDS) и межгенных спейсерах (IGS). Результаты показали, что нуклеотидное разнообразие преимущественно сосредоточено в некодирующих участках, прежде всего в межгенных спейсерах (IGS), тогда как большинство белок-кодирующих генов остаётся относительно консервативным, что согласуется с функциональными ограничениями эволюции пластидных генов. Наиболее выраженные пики Pi локализованы преимущественно в некодирующих спейсерах и сегментах, ассоциированных с областью SSC, что указывает на возможность существенного повышения эффективности маркеров путём фокусирования на нескольких наиболее информативных высоковариабельных локусах вместо широко используемых, но слабо вариабельных регионов. В качестве наиболее информативных и устойчивых по вариабельности кандидатных маркеров для Salvia определены участки ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, а также кодирующие локусы ycf1, matK, rpl16, rpl22, ndhF. В совокупности полученные данные показывают, что профилирование нуклеотидного разнообразия пластома обеспечивает надёжную, основанную на данных основу для разработки праймеров, ДНК-баркодинга и повышения точности филогенетических реконструкций Salvia, поддерживая дальнейшие таксономические и биогеографические исследования разнообразия Центральной Азии.
Филогенетикалық байланыстарды дәл анықтау және сенімді түрде қалпына келтіру көптеген өсімдік топтарында қиындық тудыруда, бұл жылдам эволюциялық сәулеленумен, айқын географиялық құрылыммен және хлоропласт геномы бойынша ақпараттық пластидтік вариацияның гетерогенді таралуымен сипатталады. Осыған байланысты, пластомдық масштабты талдаулар тығыз байланысты таксондарды зерттеген кезде дәстүрлі бір локустық маркерлерден асып түсетін филогенетикалық ақпараттық аймақтарды анықтаудың қуатты тәсілін білдіреді. Бұл зерттеуде біз пластомдық вариацияның таралуын сандық сипаттау және Орталық Азияның эндемикалық тектеріне бағытталған маркерді таңдау үшін практикалық негіз ұсыну үшін Salvia тұқымдасының хлоропласт геномының нуклеотидтік әртүрлілігін (Pi) бағаладық. Толық хлоропласт геномдары ақуызды кодтайтын гендердегі (CDS) және интергендік аралықтарда (IGS) Pi бағалау үшін сырғымалы терезе әдісін қолдана отырып тураланды және талданды. Нәтижелер нуклеотидтік әртүрліліктің негізінен кодталмайтын аймақтарда, негізінен интергендік аралықтарда (IGS) шоғырланғанын, ал ақуызды кодтайтын гендердің көпшілігі пластид генінің эволюциясындағы функционалдық шектеулерге сәйкес салыстырмалы түрде сақталғанын көрсетті. Ең көрнекті Pi шыңдары негізінен SSC аймағымен байланысты кодталмайтын спейсерлерде және сегменттерде локализацияланған, бұл кеңінен қолданылатын, бірақ өзгергіштігі төмен аймақтардың орнына ең ақпараттық, жоғары өзгермелі локустардың бірнешеуіне назар аудару арқылы маркердің өнімділігін айтарлықтай жақсарту мүмкіндігін көрсетеді. Келесі аймақтар Сальвия үшін ең ақпараттық және сенімді кандидат маркерлер ретінде анықталды: ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, сондай-ақ кодтау локустары ycf1, matK, rpl16, rpl22 және ndhF. Жалпы алғанда, алынған деректер пластом нуклеотидтерінің әртүрлілігін профильдеу праймерлерді әзірлеу, ДНҚ штрих-кодтау және Сальвия филогенетикалық реконструкцияларының дәлдігін жақсарту үшін сенімді, деректерге негізделген негізді қамтамасыз ететінін, Орталық Азия әртүрлілігін одан әрі таксономиялық және биогеографиялық зерттеулерді қолдайтынын көрсетеді.
IntroductionВведениеКіріспе
The selection of molecular markers for phylogenetic and population-level studies largely depends on accurately identifying the most variable regions of the genome. A widely used quantitative measure of sequence variability is nucleotide diversity (Pi), introduced by Tajima (1983). Pi is defined as the average number of nucleotide differences per site between all possible pairs of sequences in a sample and reflects the level of within-group genetic variation. Accordingly, Pi provides an objective criterion for comparing the informativeness of different genes and intergenic regions and enables targeted selection of candidate loci for primer development and molecular discrimination of closely related taxa.
Advances in plastome genomics have greatly expanded the toolkit for plant systematics and DNA-based identification. Analyses of complete chloroplast genomes make it possible not only to use traditional single-locus markers but also to detect genus- and lineage-specific "hotspots" of variation. Sliding-window analysis, implemented for example in DnaSP v6 (Rozas et al., 2017), is widely applied to map variability along the plastome. High Pi values indicate regions with accelerated accumulation of substitutions and thus high potential for distinguishing closely related species and, when needed, populations, whereas low Pi values reflect conserved segments with limited utility at shallow taxonomic levels. In angiosperms, plastome divergence is typically concentrated in non-coding regions, especially intergenic spacers (IGS) and introns, predominantly within the large single-copy (LSC) and small single-copy (SSC) regions, while the inverted repeats (IR) remain the most conserved.
Several studies on Salvia and other genera of Lamiaceae, largely focusing on European floras and widely distributed East Asian taxa (Nyamgerel et al., 2025; Yu et al., 2023; Gao et al., 2020; Zhao et al., 2020a), have identified a set of broadly useful variable loci suitable for primer design and molecular discrimination within the genus. For Salvia, these include trnK--rps16, rps16--trnQ, psbK--psbC, atpH--atpI, rpoB--trnC--petN, trnE--trnT, rbcL--accD, petA--psbJ, rpl16, rps3--rpl22, rpl32--trnL--ccsA, ccsA--ndhD, ndhG--ndhI, ndhA, rps15--ycf1, matK, ndhF, and ycf1 (Zhao et al., 2020b). The effectiveness of Pi-guided marker selection has also been supported by studies on taxon discrimination within several Lamiaceae genera (Shang et al., 2023; Zhao et al., 2024), demonstrating that highly variable regions provide more reliable separation of closely related species.
Nevertheless, regional, locally rare, and endemic Central Asian taxa, including those of Uzbekistan, remain underrepresented in plastome-based studies of Salvia. Expanding both geographic and taxonomic coverage can reveal additional patterns of variation and lineage-informative loci that may not be detected in datasets dominated by widely distributed species. Therefore, the aim of this study is to generate a plastome-wide nucleotide-diversity profile for Salvia and to identify the most informative candidate regions for downstream primer development, DNA barcoding, and improved phylogenetic inference of Central Asian lineages of the genus.
Materials and MethodsМатериалы и методыМатериалдар мен әдістер
Plastome Data Acquisition and Taxon Sampling
Complete chloroplast genome sequences were obtained from the National Center for Biotechnology Information (NCBI) GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). The dataset comprised representative species from Salvia across major geographic regions, with particular emphasis on Central Asian taxa to ensure adequate representation of the region endemic and locally distributed lineages. Species selection prioritized taxonomic coverage across recognized subgenera and sections, as well as phylogenetic diversity based on recent molecular studies (Wu et al., 2021; Moein et al., 2023). Only complete, annotated plastome sequences were included to ensure consistency in downstream analyses.
Sequence Alignment and Quality Control
Plastome sequences were aligned using MAFFT v7.490 (Katoh and Standley, 2013) with the FFT-NS-i iterative refinement method to accommodate length variation in intergenic regions. The alignment was manually inspected and refined in AliView v1.28 (Larsson, 2014) to correct potential misalignments, particularly in hypervariable intergenic spacers and indel-rich regions. Ambiguously aligned positions and regions with extensive gaps were flagged for interpretation but retained in the dataset to preserve information content for subsequent diversity calculations.
Nucleotide Diversity Analysis
Nucleotide diversity (Pi) was calculated using DnaSP v6.12.03 (Rozas et al., 2017), a specialized software package for analyzing DNA sequence polymorphism and estimating evolutionary parameters from multiple sequence alignments. Pi represents the average number of nucleotide differences per site between any two sequences and is computed as:
Pi = Σ xi xj πij
where xi and xj are the respective frequencies of the ith and jth sequences, and πij is the number of nucleotide differences per site between them.
A sliding-window approach was employed to profile variation continuously across the plastome. The window size was set to 600 bp with a step size of 200 bp, parameters chosen to balance regional resolution with adequate sampling of local polymorphism. These settings allow detection of localized peaks in variability while maintaining sufficient statistical power for Pi estimation within each window.
Analyses were conducted separately for protein-coding sequences (CDS) and intergenic spacers (IGS) to distinguish variation attributable to functional constraint from that arising in putatively neutral or less constrained regions. Coding sequences were extracted based on GenBank annotations, and IGS regions were defined as sequences flanked by adjacent genes or tRNAs. Introns within coding genes were analyzed as part of the IGS category. Pi values were plotted along the plastome coordinate axis to visualize variation hotspots and identify candidate loci for downstream applications.
Comparative Analysis and Marker Identification
To contextualize patterns observed in Central Asian Salvia, we performed parallel nucleotide-diversity analyses on Old World (Central Asian, Mediterranean, and East Asian) and New World taxa. Comparative profiling was conducted using identical window parameters and analytical settings to ensure methodological consistency. Regions exhibiting consistently high Pi across multiple phylogenetic groups were prioritized as candidate markers for broader applicability in Salvia systematics.
Candidate markers were selected based on multiple criteria: (1) high Pi values (>0.02) indicating substantial variation, (2) consistent performance across different Salvia lineages, (3) appropriate length for PCR amplification and Sanger sequencing (300--1500 bp), and (4) presence of conserved flanking regions suitable for universal or semi-universal primer design. Both intergenic spacers and protein-coding genes meeting these criteria were retained as recommended loci for future phylogenetic and DNA barcoding studies.
ResultsРезультатыНәтижелер
Plastome-wide Nucleotide Diversity Patterns
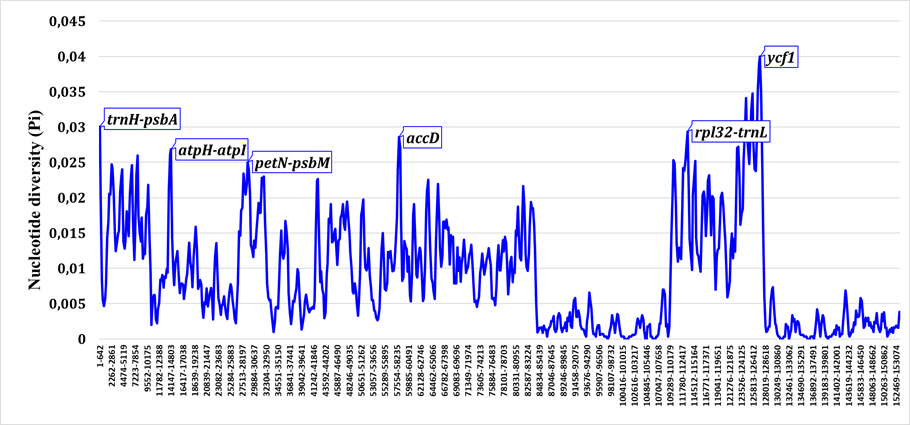
Sliding-window analysis of nucleotide diversity across the Salvia chloroplast genome revealed pronounced heterogeneity in variation levels (Fig. 1). Overall Pi values ranged from near zero in highly conserved regions to approximately 0.06 in the most variable intergenic spacers. As expected, the majority of high-variability peaks occurred in non-coding regions, particularly intergenic spacers (IGS), while protein-coding sequences (CDS) exhibited substantially lower diversity consistent with purifying selection.
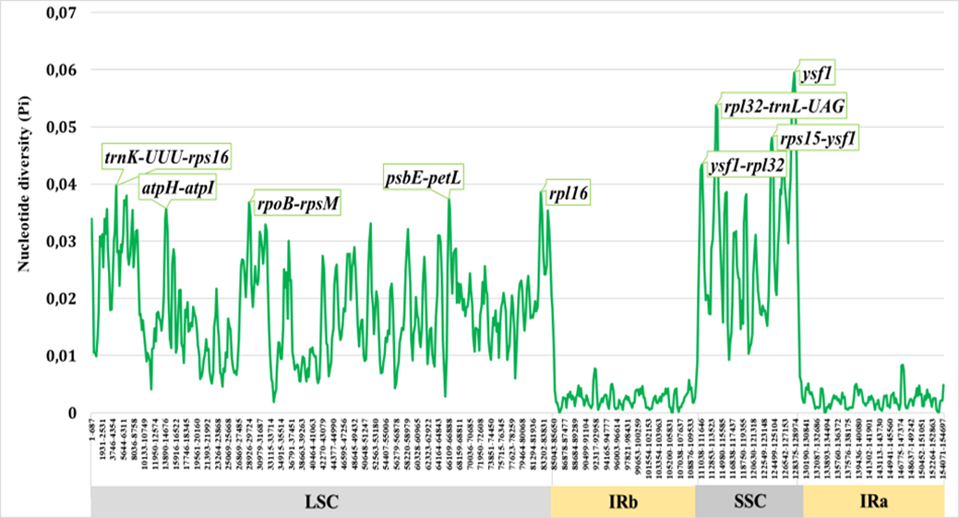
Nucleotide diversity (Pi) across the complete chloroplast genome of Salvia calculated using a sliding-window approach (window size: 600 bp, step size: 200 bp). The x-axis represents the position along the plastome, and the y-axis represents Pi values. Major variability hotspots are labeled.
Distribution of Variation in Coding vs. Non-coding Regions
When analyzed separately, intergenic spacers (IGS) showed mean Pi values approximately 3--4 times higher than those of protein-coding regions (Fig. 2). The most variable intergenic spacers included ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, and rpl32--trnL-UAG, with Pi values reaching 0.05--0.06 in Central Asian taxa. These regions consistently outperformed traditional barcoding loci in terms of sequence variation and thus offer superior potential for species-level discrimination.
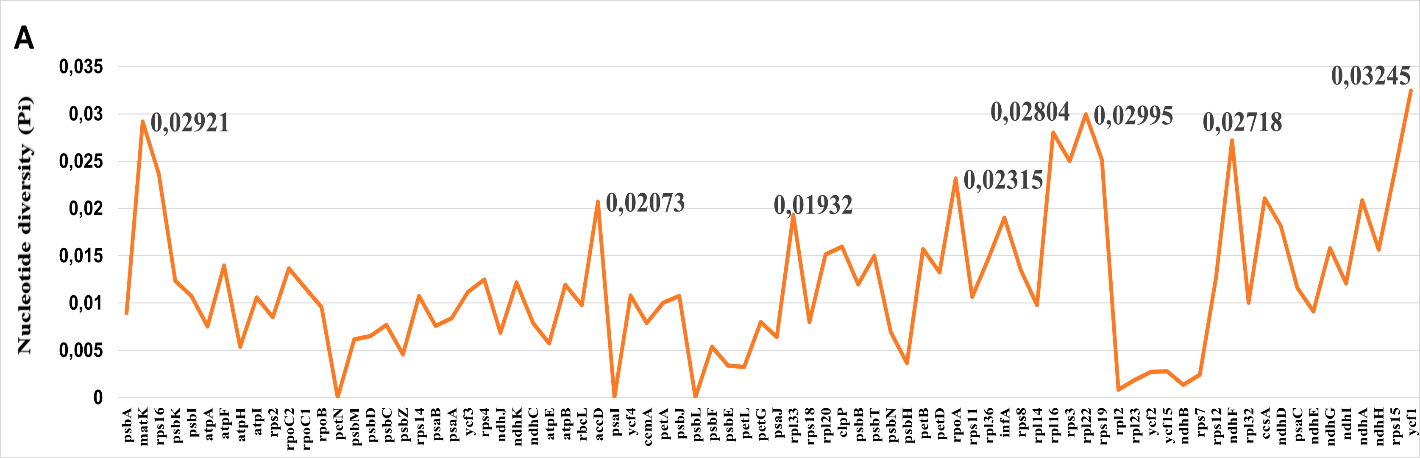
Comparison of nucleotide diversity between protein-coding sequences (CDS) and intergenic spacers (IGS) in Salvia plastomes. IGS regions exhibit significantly higher variation consistent with relaxed functional constraints.
Among protein-coding genes, ycf1, matK, rpl16, rpl22, and ndhF displayed the highest nucleotide diversity (Pi = 0.015--0.025). The gene ycf1, in particular, exhibited elevated polymorphism across its entire length, making it a strong candidate for phylogenetic studies at multiple taxonomic levels. In contrast, photosystem genes (psa and psb families) and ribosomal proteins remained highly conserved (Pi < 0.005), reflecting strong purifying selection associated with their essential cellular functions.
Regional Hotspots of Chloroplast Variation
High-variability hotspots were not uniformly distributed across the plastome but instead clustered in specific regions (Fig. 3). The small single-copy (SSC) region exhibited disproportionately high diversity relative to its size, with several intergenic spacers (e.g., ccsA--ndhD, ndhG--ndhI) and the terminal portion of ycf1 contributing substantially to overall plastome variation. Within the large single-copy (LSC) region, hotspots were associated primarily with intergenic spacers located between tRNA genes and functionally unrelated protein-coding loci.
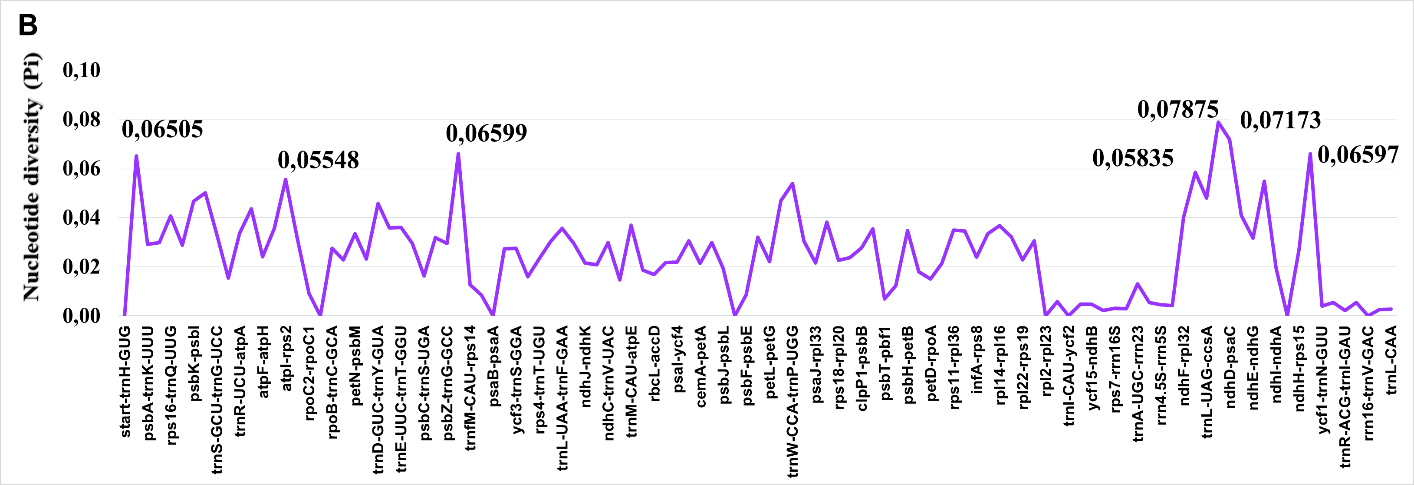
Regional distribution of nucleotide diversity hotspots across the Salvia chloroplast genome. The SSC region shows disproportionately high variation, while inverted repeats (IRa and IRb) remain highly conserved.
Comparative Analysis: Old World vs. New World Salvia
Comparative nucleotide-diversity profiling revealed substantial differences between Old World (including Central Asian) and New World Salvia lineages (Fig. 4, Fig. 5). Old World taxa, which represent evolutionarily older lineages, exhibited higher overall plastome variability (maximum Pi ≈ 0.06) compared to New World taxa (maximum Pi ≈ 0.04). Despite these differences in magnitude, the same loci (trnH--psbA, atpH--atpI, accD, rpl32--trnL, ycf1) emerged as variability peaks in both groups, indicating that these regions are consistently informative across Salvia regardless of geographic origin.
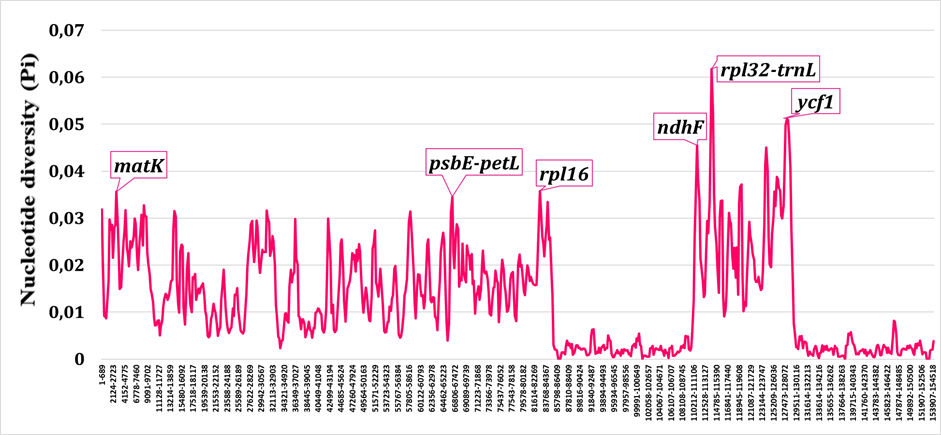
Nucleotide diversity profile for Old World Salvia taxa (Central Asian, Mediterranean, and East Asian lineages). Higher overall Pi values reflect greater evolutionary age and time for mutation accumulation.
Nucleotide diversity profile for New World Salvia taxa (primarily subgenus Calosphace). Lower Pi values reflect more recent diversification and less time for mutational accumulation compared to Old World lineages.
DiscussionОбсуждениеТалқылау
Plastome Evolution and Nucleotide Diversity Patterns
Our results demonstrate that nucleotide diversity in the Salvia chloroplast genome is strongly partitioned between coding and non-coding regions, with intergenic spacers exhibiting substantially higher variation than protein-coding sequences. This pattern is consistent with expectations based on functional constraint: coding regions, particularly those encoding photosynthetic and ribosomal proteins, are under strong purifying selection to maintain protein function, whereas intergenic spacers experience relaxed constraint and accumulate substitutions more freely (Kelchner, 2000; Dong et al., 2012).
The elevated diversity observed in the SSC region relative to the LSC and IR regions has been documented in other angiosperm lineages and likely reflects both the absence of stabilizing selection typical of IR-mediated gene conversion and the presence of genes (ndh complex, ycf1) that are less functionally constrained than photosystem components. The gene ycf1, which encodes a large protein involved in protein translocation, has been widely recognized as one of the most variable plastid loci and has proven useful for species-level phylogenetics across multiple plant families, including Lamiaceae (Chen et al., 2023).
Evolutionary History and Geographic Context
The observed differences in nucleotide diversity between Old World and New World Salvia are best explained by differences in evolutionary age and diversification history. Old World lineages, particularly those from Central Asia and the Mediterranean, represent older clades that have had more time to accumulate plastome variation. Phylogenetic and biogeographic analyses (e.g., Chen et al., 2022; Moein et al., 2023) indicate that Salvia originated in the Old World, likely in the Irano-Turanian region, during the Oligocene to early Miocene. These lineages subsequently underwent extensive diversification across temperate Eurasia and the Mediterranean, providing ample time for mutation accumulation.
In contrast, New World Salvia, particularly the large subgenus Calosphace, represents a much younger radiation. Molecular dating studies (Kriebel et al., 2019) estimate that the major diversification of Calosphace occurred around 15 Ma, with dispersal from Mexico into Central and South America beginning no earlier than ~12 Ma and continuing into the Late Miocene and Pliocene. Consequently, New World lineages have had approximately half the time available for plastome divergence compared to their Old World relatives, which is reflected in the lower maximum Pi values observed in our analyses.
Additionally, environmental factors may have influenced the rate of plastome evolution. Central Asian Salvia species often inhabit arid and semi-arid environments characterized by strong climatic seasonality and variable water availability. Such conditions may have promoted adaptive diversification and potentially accelerated the fixation of plastid mutations linked to photosynthetic efficiency or stress tolerance, though direct evidence for adaptive plastome evolution in Salvia remains limited.
Implications for Marker Selection and Phylogenetic Inference
The identification of high-variability loci provides a clear, data-driven basis for selecting molecular markers tailored to specific phylogenetic or population-genetic questions. For species-level phylogenetics and DNA barcoding within Central Asian Salvia, the intergenic spacers ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, and rpl32--trnL-UAG offer superior resolution compared to traditional single-locus barcodes such as matK or rbcL. These regions not only exhibit high Pi values but also show consistent variability across different Salvia lineages, making them broadly applicable for molecular discrimination.
Among protein-coding genes, ycf1, matK, rpl16, rpl22, and ndhF represent the best candidates for phylogenetic reconstruction at deeper taxonomic levels (e.g., subgenus or section). The gene ycf1, in particular, combines high variability with sufficient length (~5--6 kb) to provide robust phylogenetic signal and has been successfully employed in studies of Lamiaceae phylogeny (Wu et al., 2021; Zhao et al., 2020b).
It is important to note that the informativeness of any marker depends on the evolutionary scale of the question. For shallow-level analyses (e.g., intraspecific population structure), the most variable intergenic spacers will be essential. For deeper phylogenetic questions (e.g., relationships among subgenera), moderately variable coding genes such as matK and ndhF may be more suitable, as they accumulate substitutions at a slower rate and are less prone to homoplasy.
Future Directions
While the present study provides a comprehensive plastome-wide diversity profile for Salvia, several avenues for future research remain. First, the development and empirical testing of universal or semi-universal primers targeting the high-variability loci identified here will be essential to translate these findings into practical tools for field-based studies and herbarium-based DNA barcoding initiatives. Second, integration of plastome data with nuclear markers (e.g., low-copy nuclear genes, restriction-site associated DNA sequencing) would provide a more complete picture of Salvia evolutionary history, particularly in cases where plastid introgression or incomplete lineage sorting may confound phylogenetic inference.
Third, expanding geographic and taxonomic sampling to include additional Central Asian endemics and rare taxa will improve the representativeness of diversity estimates and may reveal region-specific variability hotspots not detected in the present analysis. Finally, population-level sampling of selected species across environmental gradients would enable investigation of potential adaptive signals in plastome variation, complementing the neutral evolutionary processes that predominantly shape chloroplast genome evolution.
ConclusionsВыводыҚорытынды
In summary, our nucleotide-diversity profiling of the Salvia chloroplast genome reveals that variation is concentrated in intergenic spacers and a subset of protein-coding genes, with substantial differences in overall diversity between Old World and New World lineages reflecting their differing evolutionary ages. The high-variability loci identified here—ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, ycf1, matK, rpl16, rpl22, and ndhF—provide a robust, empirically validated foundation for primer development, DNA barcoding, and improved phylogenetic inference in Salvia. These findings support future taxonomic, biogeographic, and conservation studies of Central Asian plant diversity and demonstrate the value of plastome-scale analyses for optimizing marker selection in complex and rapidly evolving plant groups.
Data & Code AvailabilityДоступность данных и кодаДеректер мен кодқа қолжетімділік
Chloroplast genome sequences used in this study are publicly available from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). Alignment files and nucleotide diversity profiles are available upon request from the corresponding author.
AcknowledgmentsБлагодарностиАлғыс сөздер
This research was supported by the State Program "Digital Nature: Development of a digital platform for the flora of Central Uzbekistan" implemented by the Institute of Botany of the Academy of Sciences of the Republic of Uzbekistan for the period 2025--2029. This research was also supported by the project titled "Assessing climate change adaptation in endangered plants of Uzbekistan: A DNA barcoding approach" (AL 9224104464). Additional support was provided by the project titled "Molecular-genetic identification of medicinal plant species in the flora of Uzbekistan and Belarus using DNA markers" (FL-7923051878).
EthicsЭтикаЭтика
Not applicable. This study used publicly available sequence data from GenBank.
Competing InterestsКонфликт интересовМүдделер қақтығысы
The authors declare no competing interests.
ReferencesЛитератураӘдебиеттер
- Chen H., Chen H., Wang B., Liu C. (2023) Conserved chloroplast genome sequences of the genus Clerodendrum Linn. (Lamiaceae) as a super-barcode. PLoS ONE, 18(2), e0277809. DOI: 10.1371/journal.pone.0277809.
- Chen Y. P., Turginov O. T., Turdimatovich T. O., Nuraliev M. S., Lazarević P., Drew B. T., Xiang C.-L. (2022) Phylogeny and biogeography of the northern temperate genus Dracocephalum s.l. (Lamiaceae). Cladistics, 38, 429--451. DOI: 10.1111/cla.12502.
- Dong W., Liu J., Yu J., Wang L., Zhou S. (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE, 7(4), e35071. DOI: 10.1371/journal.pone.0035071.
- Gao C., Wu C., Zhang Q., Zhao X., Wu M., Chen R., Zhao Y., Li Z. (2020) Characterization of chloroplast genomes from two Salvia medicinal plants and gene transfer among their mitochondrial and chloroplast genomes. Frontiers in Genetics, 11, 574962. DOI: 10.3389/fgene.2020.574962.
- Katoh K., Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772--780. DOI: 10.1093/molbev/mst010.
- Kelchner S. A. (2000) The evolution of non-coding chloroplast DNA and its application in plant systematics. Annals of the Missouri Botanical Garden, 87(4), 482--498. DOI: 10.2307/2666142.
- Kriebel R., Drew B. T., Drummond C. P., González-Gallegos J. G., Celep F., Mahdjoub M. M., Rose J. P., Xiang C.-L., Hu G.-X., Walker J. B., Lemmon E. M., Lemmon A. R., Sytsma K. J. (2019) Tracking temporal shifts in area, biomes, and pollinators in the radiation of Salvia (sages) across continents: leveraging anchored hybrid enrichment and targeted sequence data. American Journal of Botany, 106(4), 573--597. DOI: 10.1002/ajb2.1268.
- Larsson A. (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics, 30(22), 3276--3278. DOI: 10.1093/bioinformatics/btu531.
- Moein F., Jamzad Z., Rahiminejad M., et al. (2023) Towards a global perspective for Salvia L.: phylogeny, diversification and floral evolution. Journal of Evolutionary Biology, 36(3), 589--604. DOI: 10.1111/jeb.14149.
- Nyamgerel N., Baasanmunkh Sh., Munkhtulga D., Tugsbilguun T., Oyuntsetseg B., Xiang Ch.-L., Choi H. J. (2025) Characterization of the complete chloroplast genome of Dracocephalum ruyschiana (Lamiaceae) and its phylogenetic analysis. Korean Journal of Plant Taxonomy, 55(1), 44--51. DOI: 10.11110/kjpt.2025.55.1.44.
- Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J. C., et al. (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34(12), 3299--3302. DOI: 10.1093/molbev/msx248.
- Shang M., Wang J., Dai G., Zheng J., Liao B., Wang J., Duan B. (2023) Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Ajuga and common adulterants. Frontiers in Plant Science, 14, 1251829. DOI: 10.3389/fpls.2023.1251829.
- Tajima F. (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics, 105(2), 437--460. DOI: 10.1093/genetics/105.2.437.
- Wu H., Ma P. F., Li H. T., Hu G.-X., Li D. Z. (2021) Comparative plastomic analysis and insights into the phylogeny of Salvia (Lamiaceae). Plant Diversity, 43(1), 15--26. DOI: 10.1016/j.pld.2020.07.004.
- Yu D., Pei Y., Cui N., Zhao G., Hou M., Chen Y., Chen J., Li X. (2023) Comparative and phylogenetic analysis of complete chloroplast genome sequences of Salvia regarding its worldwide distribution. Scientific Reports, 13, 14268. DOI: 10.1038/s41598-023-41198-y.
- Zhao F., Li B., Drew B. T., Chen Y. P., Wang Q., Yu W. B., Liu E. D., Salmaki Y., Peng H., Xiang C. L. (2020a) Leveraging plastomes for comparative analysis and phylogenomic inference within Scutellarioideae (Lamiaceae). PLoS ONE, 15(5), e0232602. DOI: 10.1371/journal.pone.0232602.
- Zhao F., Drew B. T., Chen Y. P., Hu G. X., Li B., Xiang Ch.-L. (2020b) The chloroplast genome of Salvia: genomic characterization and phylogenetic analysis. International Journal of Plant Sciences, 181(8), 812--830. DOI: 10.1086/710083.
- Zhao Y., Chen Y. P., Drew B. T., Zhao F., Almasi M., Turginov O. T., Xiao J. F., Karimi A. G., Salmaki Y., Yu X. Q., Xiang C. L. (2024) Molecular phylogeny and taxonomy of Phlomoides (Lamiaceae, subfamily Lamioideae) in China: insights from molecular and morphological data. Plant Diversity, 46(4), 462--475. DOI: 10.1016/j.pld.2024.04.011.
Copyright © 2026 Nikitina, Ergashov, Yusupov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.