Nucleotide diversity in Central Asian Salvia L.: high-variability regions as candidate markers Нуклеотидное разнообразие центральноазиатских Salvia L.: высоковариабельные регионы как кандидатные маркеры Орталық Азиялық Salvia L. нуклеотидтік әртүрлілігі: үміткер маркерлер ретінде жоғары өзгергіштік аймақтары
AbstractАннотацияАңдатпа
Accurate species identification and reliable reconstruction of phylogenetic relationships in many plant groups remain challenging because they are often characterized by rapid evolutionary radiations, pronounced geographic structuring, and an uneven distribution of informative plastid variation across the chloroplast genome. In this context, plastome-scale analyses provide an efficient route to identify lineage-informative regions that outperform traditional single-locus markers for closely related taxa. Here we profile chloroplast nucleotide diversity (Pi) in Salvia to quantify how variation is partitioned along the chloroplast genome and to define a practical framework for marker selection targeted to Central Asian endemic lineages. Complete chloroplast genomes were aligned and analyzed using a sliding-window approach to quantify Pi across protein-coding genes (CDS) and intergenic spacers (IGS). The results show that nucleotide diversity is concentrated predominantly in non-coding regions, especially intergenic spacers, whereas most protein-coding genes remain comparatively conserved, consistent with functional constraints on plastid gene evolution. High-Pi peaks occur predominantly in non-coding spacers and SSC-associated segments, indicating that marker performance can be substantially improved by prioritizing a small panel of hotspot loci rather than relying on widely used but low-variation regions. As the most informative and consistently high-variability candidate markers for Salvia, we identified the intergenic regions ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG as well as the protein-coding loci ycf1, matK, rpl16, rpl22, ndhF. Taken together, these findings indicate that plastome nucleotide-diversity profiling offers a robust, data-driven basis for primer development, DNA barcoding, and improved phylogenetic inference in Salvia, supporting future taxonomic and biogeographic studies of Central Asian diversity.
Точная идентификация видов и надёжная реконструкция филогенетических взаимосвязей во многих группах растений остаются сложной задачей, поскольку для них характерны быстрые эволюционные радиации, выраженная географическая структурированность и неоднородное распределение информативной пластидной вариабельности по хлоропластному геному. В этом контексте пластомно-масштабные анализы представляют эффективный подход для выявления филогенетически информативных участков, превосходящих традиционные одно-локусные маркеры при исследовании близкородственных таксонов. В настоящей работе мы оценили нуклеотидное разнообразие (Pi) хлоропластного генома рода Salvia, чтобы количественно охарактеризовать распределение вариаций пластома и предложить практическую основу отбора маркеров, ориентированную на эндемичные линии Центральной Азии. Полные хлоропластные геномы были выровнены и проанализированы методом скользящего окна для оценки Pi в белок-кодирующих генах (CDS) и межгенных спейсерах (IGS). Результаты показали, что нуклеотидное разнообразие преимущественно сосредоточено в некодирующих участках, прежде всего в межгенных спейсерах (IGS), тогда как большинство белок-кодирующих генов остаётся относительно консервативным, что согласуется с функциональными ограничениями эволюции пластидных генов. Наиболее выраженные пики Pi локализованы преимущественно в некодирующих спейсерах и сегментах, ассоциированных с областью SSC, что указывает на возможность существенного повышения эффективности маркеров путём фокусирования на нескольких наиболее информативных высоковариабельных локусах вместо широко используемых, но слабо вариабельных регионов. В качестве наиболее информативных и устойчивых по вариабельности кандидатных маркеров для Salvia определены участки ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, а также кодирующие локусы ycf1, matK, rpl16, rpl22, ndhF. В совокупности полученные данные показывают, что профилирование нуклеотидного разнообразия пластома обеспечивает надёжную, основанную на данных основу для разработки праймеров, ДНК-баркодинга и повышения точности филогенетических реконструкций Salvia, поддерживая дальнейшие таксономические и биогеографические исследования разнообразия Центральной Азии.
Филогенетикалық байланыстарды дәл анықтау және сенімді түрде қалпына келтіру көптеген өсімдік топтарында қиындық тудыруда, бұл жылдам эволюциялық сәулеленумен, айқын географиялық құрылыммен және хлоропласт геномы бойынша ақпараттық пластидтік вариацияның гетерогенді таралуымен сипатталады. Осыған байланысты, пластомдық масштабты талдаулар тығыз байланысты таксондарды зерттеген кезде дәстүрлі бір локустық маркерлерден асып түсетін филогенетикалық ақпараттық аймақтарды анықтаудың қуатты тәсілін білдіреді. Бұл зерттеуде біз пластомдық вариацияның таралуын сандық сипаттау және Орталық Азияның эндемикалық тектеріне бағытталған маркерді таңдау үшін практикалық негіз ұсыну үшін Salvia тұқымдасының хлоропласт геномының нуклеотидтік әртүрлілігін (Pi) бағаладық. Толық хлоропласт геномдары ақуызды кодтайтын гендердегі (CDS) және интергендік аралықтарда (IGS) Pi бағалау үшін сырғымалы терезе әдісін қолдана отырып тураланды және талданды. Нәтижелер нуклеотидтік әртүрліліктің негізінен кодталмайтын аймақтарда, негізінен интергендік аралықтарда (IGS) шоғырланғанын, ал ақуызды кодтайтын гендердің көпшілігі пластид генінің эволюциясындағы функционалдық шектеулерге сәйкес салыстырмалы түрде сақталғанын көрсетті. Ең көрнекті Pi шыңдары негізінен SSC аймағымен байланысты кодталмайтын спейсерлерде және сегменттерде локализацияланған, бұл кеңінен қолданылатын, бірақ өзгергіштігі төмен аймақтардың орнына ең ақпараттық, жоғары өзгермелі локустардың бірнешеуіне назар аудару арқылы маркердің өнімділігін айтарлықтай жақсарту мүмкіндігін көрсетеді. Келесі аймақтар Сальвия үшін ең ақпараттық және сенімді кандидат маркерлер ретінде анықталды: ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, сондай-ақ кодтау локустары ycf1, matK, rpl16, rpl22 және ndhF. Жалпы алғанда, алынған деректер пластом нуклеотидтерінің әртүрлілігін профильдеу праймерлерді әзірлеу, ДНҚ штрих-кодтау және Сальвия филогенетикалық реконструкцияларының дәлдігін жақсарту үшін сенімді, деректерге негізделген негізді қамтамасыз ететінін, Орталық Азия әртүрлілігін одан әрі таксономиялық және биогеографиялық зерттеулерді қолдайтынын көрсетеді.
IntroductionВведениеКіріспе
The selection of molecular markers for phylogenetic and population-level studies largely depends on accurately identifying the most variable regions of the genome. A widely used quantitative measure of sequence variability is nucleotide diversity (Pi), introduced by Tajima (1983). Pi is defined as the average number of nucleotide differences per site between all possible pairs of sequences in a sample and reflects the level of within-group genetic variation. Accordingly, Pi provides an objective criterion for comparing the informativeness of different genes and intergenic regions and enables targeted selection of candidate loci for primer development and molecular discrimination of closely related taxa.Выбор молекулярных маркеров для филогенетических и популяционно-генетических исследований во многом зависит от точного выявления наиболее изменчивых участков генома. Широко используемой количественной мерой изменчивости последовательностей является нуклеотидное разнообразие (Pi), введённое Tajima (1983). Pi определяется как среднее число нуклеотидных различий на сайт между всеми возможными парами последовательностей в выборке и отражает уровень внутригрупповой генетической изменчивости. Соответственно, Pi предоставляет объективный критерий для сравнения информативности различных генов и межгенных областей и позволяет проводить целенаправленный отбор локусов-кандидатов для разработки праймеров и молекулярного различения близкородственных таксонов.Филогенетикалық және популяциялық-генетикалық зерттеулерге арналған молекулалық маркерлерді таңдау геномның ең өзгергіш аймақтарын дәл анықтауға айтарлықтай байланысты. Тізбек өзгергіштігінің кеңінен қолданылатын сандық өлшемі — Tajima (1983) енгізген нуклеотидтік әртүрлілік (Pi). Pi выборкадағы тізбектердің барлық мүмкін жұптары арасындағы бір сайтқа шаққандағы нуклеотидтік айырмашылықтардың орташа саны ретінде анықталады және топ ішілік генетикалық өзгергіштік деңгейін көрсетеді. Сәйкесінше, Pi әртүрлі гендер мен межгендік аймақтардың ақпараттылығын салыстыруға арналған объективті критерий ұсынады және праймерлерді әзірлеу мен тығыз туыс таксондарды молекулалық ажырату үшін локус-кандидаттарды мақсатты түрде іріктеуге мүмкіндік береді.
Advances in plastome genomics have greatly expanded the toolkit for plant systematics and DNA-based identification. Analyses of complete chloroplast genomes make it possible not only to use traditional single-locus markers but also to detect genus- and lineage-specific "hotspots" of variation. Sliding-window analysis, implemented for example in DnaSP v6 (Rozas et al., 2017), is widely applied to map variability along the plastome. High Pi values indicate regions with accelerated accumulation of substitutions and thus high potential for distinguishing closely related species and, when needed, populations, whereas low Pi values reflect conserved segments with limited utility at shallow taxonomic levels. In angiosperms, plastome divergence is typically concentrated in non-coding regions, especially intergenic spacers (IGS) and introns, predominantly within the large single-copy (LSC) and small single-copy (SSC) regions, while the inverted repeats (IR) remain the most conserved.Достижения геномики пластомов значительно расширили инструментарий систематики растений и идентификации на основе ДНК. Анализ полных хлоропластных геномов позволяет не только использовать традиционные однолокусные маркеры, но и выявлять родо- и линиеспецифичные «горячие точки» изменчивости. Анализ методом скользящего окна, реализованный, например, в DnaSP v6 (Rozas et al., 2017), широко применяется для картирования изменчивости вдоль пластома. Высокие значения Pi указывают на области с ускоренным накоплением замен и, следовательно, высоким потенциалом для различения близкородственных видов и, при необходимости, популяций, тогда как низкие значения Pi отражают консервативные сегменты с ограниченной применимостью на неглубоких таксономических уровнях. У покрытосеменных дивергенция пластома обычно сосредоточена в некодирующих областях, особенно в межгенных спейсерах (IGS) и интронах, преимущественно в пределах большой однокопийной (LSC) и малой однокопийной (SSC) областей, тогда как инвертированные повторы (IR) остаются наиболее консервативными.Пластом геномикасындағы жетістіктер өсімдіктер систематикасы мен ДНҚ негізіндегі сәйкестендіру құралдарын айтарлықтай кеңейтті. Толық хлоропласт геномдарын талдау дәстүрлі бір локусты маркерлерді қолданып қана қоймай, тұқымдасқа және тармаққа тән өзгергіштік «ыстық нүктелерін» анықтауға да мүмкіндік береді. Мысалы, DnaSP v6 (Rozas et al., 2017) бағдарламасында жүзеге асырылған сырғымалы терезе әдісі пластом бойынша өзгергіштікті картаға түсіру үшін кеңінен қолданылады. Жоғары Pi мәндері алмасулардың жеделдетілген жинақталуымен сипатталатын аймақтарды, демек тығыз туыс түрлерді және қажет болғанда популяцияларды ажыратудың жоғары әлеуетін көрсетеді, ал төмен Pi мәндері таяз таксономиялық деңгейлерде қолданылуы шектеулі консервативті сегменттерді көрсетеді. Жабық тұқымды өсімдіктерде пластом дивергенциясы әдетте кодталмайтын аймақтарда, әсіресе межгендік спейсерлерде (IGS) мен интрондарда, негізінен үлкен бір данадан тұратын (LSC) және кіші бір данадан тұратын (SSC) аймақтар шегінде шоғырланады, ал инвертталған қайталанулар (IR) ең консервативті болып қалады.
Several studies on Salvia and other genera of Lamiaceae, largely focusing on European floras and widely distributed East Asian taxa (Nyamgerel et al., 2025; Yu et al., 2023; Gao et al., 2020; Zhao et al., 2020a), have identified a set of broadly useful variable loci suitable for primer design and molecular discrimination within the genus. For Salvia, these include trnK--rps16, rps16--trnQ, psbK--psbC, atpH--atpI, rpoB--trnC--petN, trnE--trnT, rbcL--accD, petA--psbJ, rpl16, rps3--rpl22, rpl32--trnL--ccsA, ccsA--ndhD, ndhG--ndhI, ndhA, rps15--ycf1, matK, ndhF, and ycf1 (Zhao et al., 2020b). The effectiveness of Pi-guided marker selection has also been supported by studies on taxon discrimination within several Lamiaceae genera (Shang et al., 2023; Zhao et al., 2024), demonstrating that highly variable regions provide more reliable separation of closely related species.В ряде работ по Salvia и другим родам Lamiaceae, в значительной мере сосредоточенных на флорах Европы и широко распространённых восточноазиатских таксонах (Nyamgerel et al., 2025; Yu et al., 2023; Gao et al., 2020; Zhao et al., 2020a), был выявлен набор широко применимых изменчивых локусов, пригодных для конструирования праймеров и молекулярного различения внутри рода. Для Salvia к ним относятся trnK--rps16, rps16--trnQ, psbK--psbC, atpH--atpI, rpoB--trnC--petN, trnE--trnT, rbcL--accD, petA--psbJ, rpl16, rps3--rpl22, rpl32--trnL--ccsA, ccsA--ndhD, ndhG--ndhI, ndhA, rps15--ycf1, matK, ndhF и ycf1 (Zhao et al., 2020b). Эффективность отбора маркеров на основе Pi также подтверждена исследованиями по различению таксонов внутри нескольких родов Lamiaceae (Shang et al., 2023; Zhao et al., 2024), показавшими, что высокоизменчивые области обеспечивают более надёжное разделение близкородственных видов.Salvia және Lamiaceae тұқымдасының басқа да туыстары бойынша негізінен Еуропа флораларына және кеңінен таралған шығысазиялық таксондарға бағытталған бірқатар зерттеулер (Nyamgerel et al., 2025; Yu et al., 2023; Gao et al., 2020; Zhao et al., 2020a) праймерлерді жобалауға және тұқымдас ішінде молекулалық ажыратуға жарамды кеңінен қолданылатын өзгергіш локустар жиынтығын анықтады. Salvia үшін оларға trnK--rps16, rps16--trnQ, psbK--psbC, atpH--atpI, rpoB--trnC--petN, trnE--trnT, rbcL--accD, petA--psbJ, rpl16, rps3--rpl22, rpl32--trnL--ccsA, ccsA--ndhD, ndhG--ndhI, ndhA, rps15--ycf1, matK, ndhF және ycf1 жатады (Zhao et al., 2020b). Pi негізіндегі маркерлерді іріктеудің тиімділігі Lamiaceae бірнеше туысы ішінде таксондарды ажырату жөніндегі зерттеулермен де расталды (Shang et al., 2023; Zhao et al., 2024), бұл жоғары өзгергіш аймақтар тығыз туыс түрлерді сенімдірек бөлуді қамтамасыз ететінін көрсетеді.
Nevertheless, regional, locally rare, and endemic Central Asian taxa, including those of Uzbekistan, remain underrepresented in plastome-based studies of Salvia. Expanding both geographic and taxonomic coverage can reveal additional patterns of variation and lineage-informative loci that may not be detected in datasets dominated by widely distributed species. Therefore, the aim of this study is to generate a plastome-wide nucleotide-diversity profile for Salvia and to identify the most informative candidate regions for downstream primer development, DNA barcoding, and improved phylogenetic inference of Central Asian lineages of the genus.Тем не менее региональные, локально редкие и эндемичные центральноазиатские таксоны, в том числе таксоны Узбекистана, остаются недостаточно представленными в основанных на пластомах исследованиях Salvia. Расширение как географического, так и таксономического охвата может выявить дополнительные закономерности изменчивости и линиеинформативные локусы, которые могут оставаться незамеченными в наборах данных, где преобладают широко распространённые виды. Поэтому цель настоящего исследования — построить профиль нуклеотидного разнообразия в масштабе всего пластома для Salvia и выявить наиболее информативные области-кандидаты для последующей разработки праймеров, ДНК-штрихкодирования и повышения точности филогенетического анализа центральноазиатских линий рода.Дегенмен Өзбекстан таксондарын қоса алғанда, аймақтық, жергілікті сирек кездесетін және эндемик Орталық Азия таксондары Salvia пластомына негізделген зерттеулерде әлі де жеткіліксіз ұсынылған. Географиялық та, таксономиялық та қамтуды кеңейту кеңінен таралған түрлер басым деректер жиынтығында анықталмауы мүмкін өзгергіштіктің қосымша заңдылықтары мен тармаққа ақпараттық локустарды аша алады. Сондықтан осы зерттеудің мақсаты — Salvia үшін бүкіл пластом ауқымындағы нуклеотидтік әртүрлілік профилін құру және тұқымдастың Орталық Азия тармақтарын кейінгі праймер әзірлеу, ДНҚ-штрихкодтау және филогенетикалық тұжырымды жақсарту үшін ең ақпараттық үміткер аймақтарды анықтау.
Materials and MethodsМатериалы и методыМатериалдар мен әдістер
Plastome Data Acquisition and Taxon SamplingПолучение данных пластомов и отбор таксоновПластом деректерін алу және таксондарды іріктеу
Complete chloroplast genome sequences were obtained from the National Center for Biotechnology Information (NCBI) GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). The dataset comprised representative species from Salvia across major geographic regions, with particular emphasis on Central Asian taxa to ensure adequate representation of the region endemic and locally distributed lineages. Species selection prioritized taxonomic coverage across recognized subgenera and sections, as well as phylogenetic diversity based on recent molecular studies (Wu et al., 2021; Moein et al., 2023). Only complete, annotated plastome sequences were included to ensure consistency in downstream analyses.Полные последовательности хлоропластных геномов были получены из базы данных GenBank Национального центра биотехнологической информации (NCBI) (https://www.ncbi.nlm.nih.gov/genbank/). Набор данных включал репрезентативные виды рода Salvia из основных географических регионов с особым акцентом на центральноазиатские таксоны, что обеспечивало достаточную представленность эндемичных и локально распространённых линий региона. При отборе видов приоритет отдавался таксономическому охвату признанных подродов и секций, а также филогенетическому разнообразию на основе современных молекулярных исследований (Wu et al., 2021; Moein et al., 2023). В анализ включали только полные аннотированные последовательности пластомов, что обеспечивало согласованность последующих аналитических процедур.Хлоропласт геномдарының толық тізбектері Биотехнологиялық ақпараттың ұлттық орталығының (NCBI) GenBank дерекқорынан алынды (https://www.ncbi.nlm.nih.gov/genbank/). Деректер жиынтығы негізгі географиялық аймақтардағы Salvia тұқымдасының репрезентативті түрлерін қамтыды, әсіресе аймақтың эндемик және жергілікті таралған тұқымдарының жеткілікті түрде ұсынылуын қамтамасыз ету үшін Орталық Азия таксондарына ерекше назар аударылды. Түрлерді іріктеу кезінде танылған тұқымдас ішілік топтар мен секцияларды таксономиялық қамту, сондай-ақ заманауи молекулалық зерттеулерге негізделген филогенетикалық әртүрлілік басымдыққа ие болды (Wu et al., 2021; Moein et al., 2023). Кейінгі талдаулардың дәйектілігін қамтамасыз ету үшін тек толық, аннотацияланған пластом тізбектері ғана қосылды.
Sequence Alignment and Quality ControlВыравнивание последовательностей и контроль качестваТізбектерді туралау және сапаны бақылау
Plastome sequences were aligned using MAFFT v7.490 (Katoh and Standley, 2013) with the FFT-NS-i iterative refinement method to accommodate length variation in intergenic regions. The alignment was manually inspected and refined in AliView v1.28 (Larsson, 2014) to correct potential misalignments, particularly in hypervariable intergenic spacers and indel-rich regions. Ambiguously aligned positions and regions with extensive gaps were flagged for interpretation but retained in the dataset to preserve information content for subsequent diversity calculations.Последовательности пластомов выравнивали с помощью MAFFT v7.490 (Katoh and Standley, 2013) с использованием метода итеративного уточнения FFT-NS-i для учёта различий в длине межгенных областей. Выравнивание проверяли вручную и дорабатывали в AliView v1.28 (Larsson, 2014) для исправления возможных ошибок выравнивания, особенно в гипервариабельных межгенных спейсерах и областях, богатых инделями. Позиции с неоднозначным выравниванием и области с протяжёнными пропусками отмечали для последующей интерпретации, но сохраняли в наборе данных, чтобы не утратить информативность при дальнейших расчётах разнообразия.Пластом тізбектері межгендік аймақтардағы ұзындық айырмашылықтарын ескеру үшін FFT-NS-i итеративті нақтылау әдісімен MAFFT v7.490 (Katoh and Standley, 2013) бағдарламасы арқылы тураланды. Туралау AliView v1.28 (Larsson, 2014) бағдарламасында қолмен тексеріліп, әсіресе гипервариабельді межгендік спейсерлерде және индельдерге бай аймақтарда ықтимал туралау қателерін түзету мақсатында нақтыланды. Туралауы анық емес позициялар мен ұзын саңылаулары бар аймақтар интерпретация үшін белгіленді, бірақ кейінгі әртүрлілік есептеулерінің ақпараттық мазмұнын сақтау үшін деректер жиынтығында қалдырылды.
Nucleotide Diversity AnalysisАнализ нуклеотидного разнообразияНуклеотидтік әртүрлілікті талдау
Nucleotide diversity (Pi) was calculated using DnaSP v6.12.03 (Rozas et al., 2017), a specialized software package for analyzing DNA sequence polymorphism and estimating evolutionary parameters from multiple sequence alignments. Pi represents the average number of nucleotide differences per site between any two sequences and is computed as:Нуклеотидное разнообразие (Pi) рассчитывали с помощью DnaSP v6.12.03 (Rozas et al., 2017) — специализированного программного пакета для анализа полиморфизма последовательностей ДНК и оценки эволюционных параметров на основе множественных выравниваний. Pi представляет собой среднее число нуклеотидных различий на сайт между любыми двумя последовательностями и вычисляется как:Нуклеотидтік әртүрлілік (Pi) ДНҚ тізбектерінің полиморфизмін талдауға және көптік туралаулар негізінде эволюциялық параметрлерді бағалауға арналған мамандандырылған DnaSP v6.12.03 (Rozas et al., 2017) бағдарламалық пакеті арқылы есептелді. Pi кез келген екі тізбек арасындағы бір сайтқа шаққандағы нуклеотидтік айырмашылықтардың орташа санын білдіреді және мынадай түрде есептеледі:
Pi = Σ xi xj πij
where xi and xj are the respective frequencies of the ith and jth sequences, and πij is the number of nucleotide differences per site between them.где xi и xj — частоты i-й и j-й последовательностей соответственно, а πij — число нуклеотидных различий на сайт между ними.мұндағы xi және xj — сәйкесінше i-ші және j-ші тізбектердің жиіліктері, ал πij — олардың арасындағы бір сайтқа шаққандағы нуклеотидтік айырмашылықтардың саны.
A sliding-window approach was employed to profile variation continuously across the plastome. The window size was set to 600 bp with a step size of 200 bp, parameters chosen to balance regional resolution with adequate sampling of local polymorphism. These settings allow detection of localized peaks in variability while maintaining sufficient statistical power for Pi estimation within each window.Для непрерывного профилирования изменчивости по всему пластому применяли метод скользящего окна. Размер окна составлял 600 bp при шаге 200 bp; эти параметры выбирали для оптимального соотношения между региональным разрешением и достаточной выборкой локального полиморфизма. Такие настройки позволяют выявлять локальные пики изменчивости, сохраняя при этом достаточную статистическую мощность для оценки Pi в пределах каждого окна.Пластом бойынша өзгергіштікті үздіксіз бейіндеу үшін сырғымалы терезе әдісі қолданылды. Терезе өлшемі 600 bp, ал қадам өлшемі 200 bp болып белгіленді; бұл параметрлер аймақтық ажыратымдылық пен жергілікті полиморфизмді жеткілікті іріктеу арасындағы тепе-теңдікті қамтамасыз ету үшін таңдалды. Мұндай баптаулар әр терезе ішінде Pi бағалауының жеткілікті статистикалық қуатын сақтай отырып, өзгергіштіктің жергілікті шыңдарын анықтауға мүмкіндік береді.
Analyses were conducted separately for protein-coding sequences (CDS) and intergenic spacers (IGS) to distinguish variation attributable to functional constraint from that arising in putatively neutral or less constrained regions. Coding sequences were extracted based on GenBank annotations, and IGS regions were defined as sequences flanked by adjacent genes or tRNAs. Introns within coding genes were analyzed as part of the IGS category. Pi values were plotted along the plastome coordinate axis to visualize variation hotspots and identify candidate loci for downstream applications.Анализы проводили раздельно для белок-кодирующих последовательностей (CDS) и межгенных спейсеров (IGS), чтобы разграничить изменчивость, обусловленную функциональными ограничениями, и изменчивость, возникающую в предположительно нейтральных или менее ограниченных областях. Кодирующие последовательности извлекали на основе аннотаций GenBank, а области IGS определяли как последовательности, ограниченные соседними генами или тРНК. Интроны кодирующих генов анализировали в составе категории IGS. Значения Pi откладывали вдоль координатной оси пластома для визуализации горячих точек изменчивости и выявления локусов-кандидатов для последующего применения.Талдаулар функционалдық шектеулерден туындайтын өзгергіштікті болжамды бейтарап немесе аз шектелген аймақтарда пайда болатын өзгергіштіктен ажырату мақсатында ақуыз кодтаушы тізбектер (CDS) мен межгендік спейсерлер (IGS) үшін бөлек жүргізілді. Кодтаушы тізбектер GenBank аннотацияларына сүйеніп бөліп алынды, ал IGS аймақтары көршілес гендермен немесе тРНҚ-мен шектелген тізбектер ретінде анықталды. Кодтаушы гендердің ішіндегі интрондар IGS санатының құрамында талданды. Pi мәндері өзгергіштіктің ыстық нүктелерін көрнекілендіру және кейінгі қолданысқа арналған локус-кандидаттарды анықтау үшін пластомның координаталық осі бойымен сызылды.
Comparative Analysis and Marker IdentificationСравнительный анализ и идентификация маркеровСалыстырмалы талдау және маркерлерді анықтау
To contextualize patterns observed in Central Asian Salvia, we performed parallel nucleotide-diversity analyses on Old World (Central Asian, Mediterranean, and East Asian) and New World taxa. Comparative profiling was conducted using identical window parameters and analytical settings to ensure methodological consistency. Regions exhibiting consistently high Pi across multiple phylogenetic groups were prioritized as candidate markers for broader applicability in Salvia systematics.Чтобы поместить выявленные у центральноазиатских Salvia закономерности в более широкий контекст, мы провели параллельные анализы нуклеотидного разнообразия для таксонов Старого Света (центральноазиатских, средиземноморских и восточноазиатских) и Нового Света. Сравнительное профилирование выполняли с использованием идентичных параметров окна и аналитических настроек для обеспечения методической согласованности. Области, стабильно демонстрировавшие высокие значения Pi в нескольких филогенетических группах, рассматривали в первую очередь как маркеры-кандидаты с более широкой применимостью в систематике Salvia.Орталық Азия Salvia өкілдерінде байқалған заңдылықтарды кеңірек контекстке орналастыру үшін біз Ескі Дүние (орталықазиялық, жерорта теңіздік және шығысазиялық) және Жаңа Дүние таксондары үшін параллель нуклеотидтік әртүрлілік талдауларын жүргіздік. Салыстырмалы бейіндеу әдістемелік дәйектілікті қамтамасыз ету мақсатында бірдей терезе параметрлері мен талдау баптаулары арқылы орындалды. Бірнеше филогенетикалық топта тұрақты түрде жоғары Pi мәндерін көрсеткен аймақтар Salvia систематикасында кеңірек қолданысқа ие маркер-кандидаттар ретінде басымдыққа алынды.
Candidate markers were selected based on multiple criteria: (1) high Pi values (>0.02) indicating substantial variation, (2) consistent performance across different Salvia lineages, (3) appropriate length for PCR amplification and Sanger sequencing (300--1500 bp), and (4) presence of conserved flanking regions suitable for universal or semi-universal primer design. Both intergenic spacers and protein-coding genes meeting these criteria were retained as recommended loci for future phylogenetic and DNA barcoding studies.Маркеры-кандидаты отбирали по нескольким критериям: (1) высокие значения Pi (>0.02), указывающие на значительную изменчивость; (2) стабильная информативность у разных линий Salvia; (3) подходящая длина для ПЦР-амплификации и секвенирования по Сэнгеру (300--1500 bp); (4) наличие консервативных фланкирующих областей, пригодных для конструирования универсальных или полууниверсальных праймеров. Как межгенные спейсеры, так и белок-кодирующие гены, удовлетворявшие этим критериям, были сохранены в качестве рекомендуемых локусов для будущих филогенетических исследований и ДНК-штрихкодирования.Маркер-кандидаттар бірнеше критерий бойынша іріктелді: (1) айтарлықтай өзгергіштікті көрсететін жоғары Pi мәндері (>0.02); (2) әртүрлі Salvia тұқымдарында тұрақты ақпараттылық; (3) ПТР-амплификация мен Сэнгер бойынша секвенирлеуге қолайлы ұзындық (300--1500 bp); (4) әмбебап немесе жартылай әмбебап праймерлерді жобалауға жарамды консервативті жанама аймақтардың болуы. Осы критерийлерге сай келген межгендік спейсерлер де, ақуыз кодтаушы гендер де болашақ филогенетикалық зерттеулер мен ДНҚ-штрихкодтау үшін ұсынылатын локустар ретінде сақталды.
ResultsРезультатыНәтижелер
Plastome-wide Nucleotide Diversity PatternsЗакономерности нуклеотидного разнообразия в масштабе всего пластомаПластом ауқымындағы нуклеотидтік әртүрлілік заңдылықтары
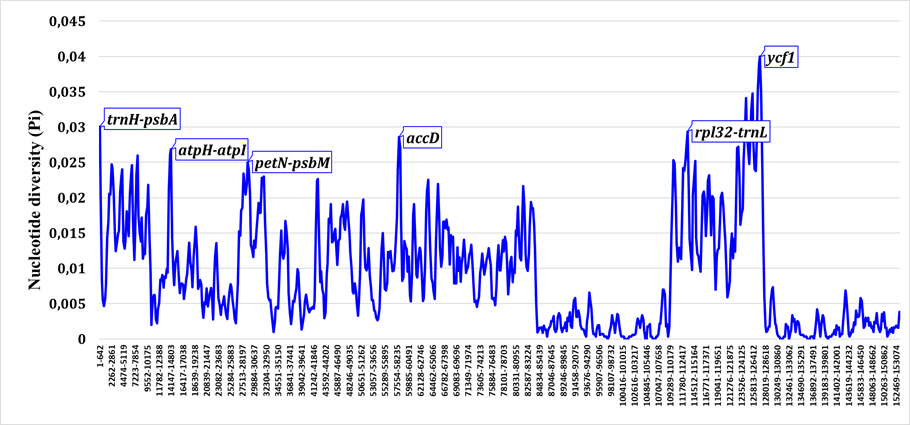
Sliding-window analysis of nucleotide diversity across the Salvia chloroplast genome revealed pronounced heterogeneity in variation levels (Fig. 1). Overall Pi values ranged from near zero in highly conserved regions to approximately 0.06 in the most variable intergenic spacers. As expected, the majority of high-variability peaks occurred in non-coding regions, particularly intergenic spacers (IGS), while protein-coding sequences (CDS) exhibited substantially lower diversity consistent with purifying selection.Анализ нуклеотидного разнообразия методом скользящего окна по хлоропластному геному Salvia выявил выраженную неоднородность уровней изменчивости (Fig. 1). Значения Pi варьировали от близких к нулю в высококонсервативных участках до приблизительно 0.06 в наиболее изменчивых межгенных спейсерах. Как и ожидалось, большинство пиков высокой изменчивости приходилось на некодирующие участки, в особенности межгенные спейсеры (IGS), тогда как белок-кодирующие последовательности (CDS) демонстрировали существенно более низкое разнообразие, что согласуется с действием очищающего отбора.Сырғымалы терезе әдісімен Salvia хлоропласт геномы бойынша жүргізілген нуклеотидтік әртүрлілік талдауы өзгергіштік деңгейлерінің айқын біркелкі еместігін көрсетті (Fig. 1). Жалпы Pi мәндері жоғары консервативті аймақтарда нөлге жуық мәннен ең өзгергіш межгендік спейсерлерде шамамен 0.06 дейін ауытқыды. Күтілгендей, жоғары өзгергіштік шыңдарының басым бөлігі кодталмайтын аймақтарда, әсіресе межгендік спейсерлерде (IGS) орналасты, ал белок кодтайтын тізбектер (CDS) тазартушы сұрыпталуға сәйкес келетін айтарлықтай төмен әртүрлілік көрсетті.
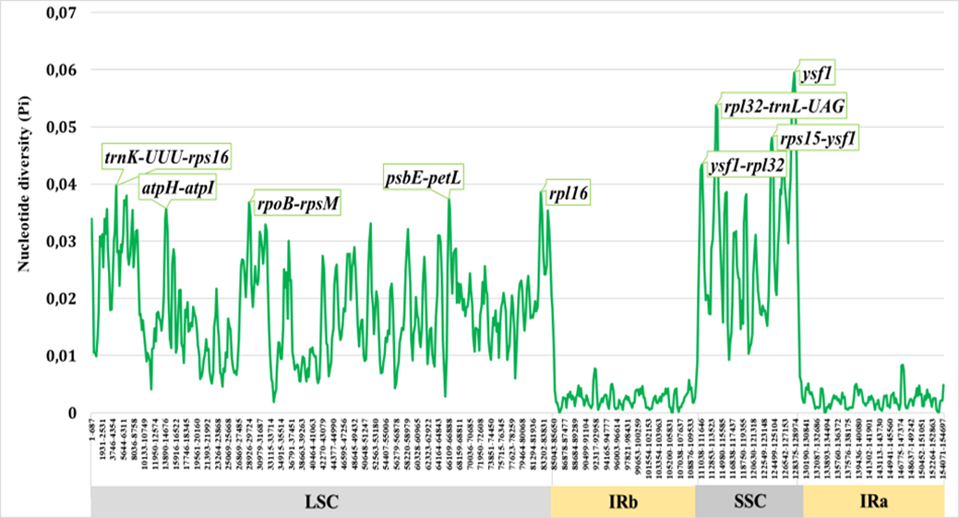
Distribution of Variation in Coding vs. Non-coding RegionsРаспределение изменчивости в кодирующих и некодирующих участкахКодтайтын және кодталмайтын аймақтардағы өзгергіштіктің таралуы
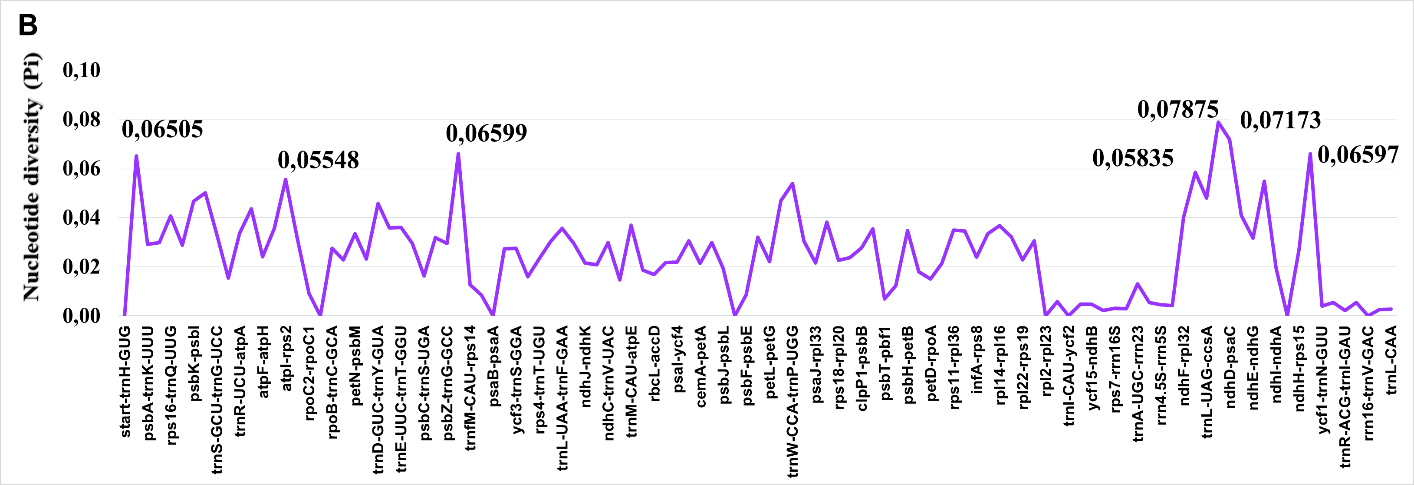
When analyzed separately, intergenic spacers (IGS) showed mean Pi values approximately 3--4 times higher than those of protein-coding regions (Fig. 2). The most variable intergenic spacers included ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, and rpl32--trnL-UAG, with Pi values reaching 0.05--0.06 in Central Asian taxa. These regions consistently outperformed traditional barcoding loci in terms of sequence variation and thus offer superior potential for species-level discrimination.При раздельном анализе межгенные спейсеры (IGS) показали средние значения Pi приблизительно в 3--4 раза выше, чем белок-кодирующие участки (Fig. 2). К наиболее изменчивым межгенным спейсерам относились ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2 и rpl32--trnL-UAG, значения Pi которых достигали 0.05--0.06 у центральноазиатских таксонов. Эти участки стабильно превосходили традиционные локусы штрихкодирования по уровню изменчивости последовательностей и тем самым обладают более высоким потенциалом для различения на видовом уровне.Бөлек талданғанда межгендік спейсерлер (IGS) орташа Pi мәндерін белок кодтайтын аймақтармен салыстырғанда шамамен 3--4 есе жоғары көрсетті (Fig. 2). Ең өзгергіш межгендік спейсерлерге ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2 және rpl32--trnL-UAG жатты, олардың Pi мәндері Орталық Азия таксондарында 0.05--0.06 жетті. Бұл аймақтар тізбек өзгергіштігі бойынша дәстүрлі штрихкодтау локустарынан тұрақты түрде асып түсті, сондықтан түр деңгейінде ажыратудың жоғары әлеуетіне ие.
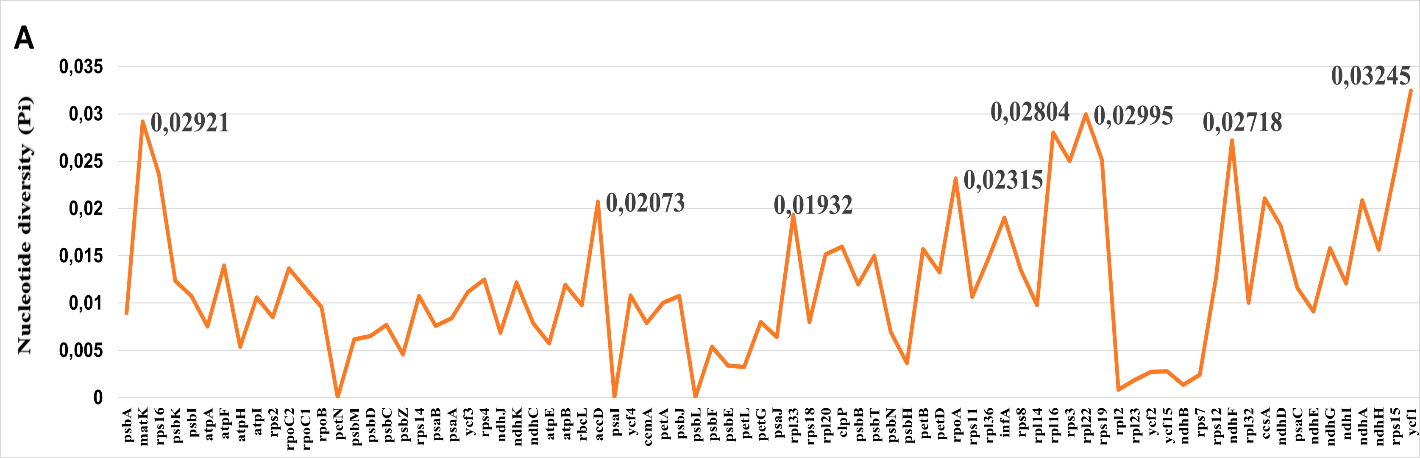
Among protein-coding genes, ycf1, matK, rpl16, rpl22, and ndhF displayed the highest nucleotide diversity (Pi = 0.015--0.025). The gene ycf1, in particular, exhibited elevated polymorphism across its entire length, making it a strong candidate for phylogenetic studies at multiple taxonomic levels. In contrast, photosystem genes (psa and psb families) and ribosomal proteins remained highly conserved (Pi < 0.005), reflecting strong purifying selection associated with their essential cellular functions.Среди белок-кодирующих генов наибольшее нуклеотидное разнообразие демонстрировали ycf1, matK, rpl16, rpl22 и ndhF (Pi = 0.015--0.025). В частности, ген ycf1 проявлял повышенный полиморфизм по всей своей длине, что делает его сильным кандидатом для филогенетических исследований на нескольких таксономических уровнях. Напротив, гены фотосистем (семейства psa и psb) и рибосомные белки оставались высококонсервативными (Pi < 0.005), что отражает сильный очищающий отбор, связанный с их жизненно важными клеточными функциями.Белок кодтайтын гендердің ішінде ең жоғары нуклеотидтік әртүрлілікті ycf1, matK, rpl16, rpl22 және ndhF көрсетті (Pi = 0.015--0.025). Атап айтқанда, ycf1 гені бүкіл ұзындығы бойынша жоғары полиморфизм көрсетті, бұл оны бірнеше таксономиялық деңгейдегі филогенетикалық зерттеулер үшін мықты үміткер етеді. Керісінше, фотожүйе гендері (psa және psb тұқымдастары) мен рибосомдық белоктар жоғары консервативті болып қалды (Pi < 0.005), бұл олардың маңызды жасушалық қызметтерімен байланысты күшті тазартушы сұрыпталуды көрсетеді.
Regional Hotspots of Chloroplast VariationРегиональные горячие точки изменчивости хлоропластаХлоропласт өзгергіштігінің аймақтық ыстық нүктелері
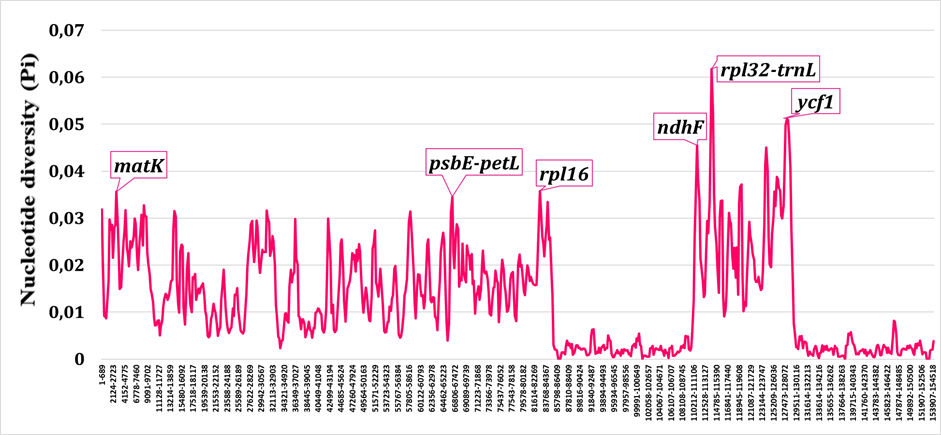
High-variability hotspots were not uniformly distributed across the plastome but instead clustered in specific regions (Fig. 3). The small single-copy (SSC) region exhibited disproportionately high diversity relative to its size, with several intergenic spacers (e.g., ccsA--ndhD, ndhG--ndhI) and the terminal portion of ycf1 contributing substantially to overall plastome variation. Within the large single-copy (LSC) region, hotspots were associated primarily with intergenic spacers located between tRNA genes and functionally unrelated protein-coding loci.Горячие точки высокой изменчивости были распределены по пластому неравномерно, а группировались в определённых участках (Fig. 3). Малая однокопийная (SSC) область демонстрировала непропорционально высокое разнообразие относительно своего размера, причём существенный вклад в общую изменчивость пластома вносили несколько межгенных спейсеров (например, ccsA--ndhD, ndhG--ndhI) и концевая часть ycf1. В пределах большой однокопийной (LSC) области горячие точки были связаны главным образом с межгенными спейсерами, расположенными между генами тРНК и функционально не связанными белок-кодирующими локусами.Жоғары өзгергіштіктің ыстық нүктелері пластом бойынша біркелкі таралмай, белгілі бір аймақтарда шоғырланды (Fig. 3). Кіші бір данадан тұратын (SSC) аймақ өз өлшеміне қатысты пропорционалды емес жоғары әртүрлілік көрсетті, мұнда пластомның жалпы өзгергіштігіне бірнеше межгендік спейсер (мысалы, ccsA--ndhD, ndhG--ndhI) және ycf1 геннің соңғы бөлігі айтарлықтай үлес қосты. Үлкен бір данадан тұратын (LSC) аймақ шегінде ыстық нүктелер негізінен тРНҚ гендері мен функционалдық байланысы жоқ белок кодтайтын локустардың арасында орналасқан межгендік спейсерлермен байланысты болды.
Comparative Analysis: Old World vs. New World SalviaСравнительный анализ: Salvia Старого и Нового СветаСалыстырмалы талдау: Ескі және Жаңа Дүние Salvia
Comparative nucleotide-diversity profiling revealed substantial differences between Old World (including Central Asian) and New World Salvia lineages (Fig. 4, Fig. 5). Old World taxa, which represent evolutionarily older lineages, exhibited higher overall plastome variability (maximum Pi ≈ 0.06) compared to New World taxa (maximum Pi ≈ 0.04). Despite these differences in magnitude, the same loci (trnH--psbA, atpH--atpI, accD, rpl32--trnL, ycf1) emerged as variability peaks in both groups, indicating that these regions are consistently informative across Salvia regardless of geographic origin.Сравнительное профилирование нуклеотидного разнообразия выявило существенные различия между линиями Salvia Старого Света (включая центральноазиатские) и Нового Света (Fig. 4, Fig. 5). Таксоны Старого Света, представляющие эволюционно более древние линии, демонстрировали более высокую общую изменчивость пластома (максимум Pi ≈ 0.06) по сравнению с таксонами Нового Света (максимум Pi ≈ 0.04). Несмотря на эти различия в величине, одни и те же локусы (trnH--psbA, atpH--atpI, accD, rpl32--trnL, ycf1) выступали в качестве пиков изменчивости в обеих группах, что указывает на стабильную информативность этих участков у Salvia независимо от географического происхождения.Нуклеотидтік әртүрлілікті салыстырмалы профильдеу Ескі Дүние (Орталық Азиялықтарды қоса алғанда) және Жаңа Дүние Salvia тұқымдық желілерінің арасында айтарлықтай айырмашылықтарды анықтады (Fig. 4, Fig. 5). Эволюциялық тұрғыдан ежелгі желілерді білдіретін Ескі Дүние таксондары Жаңа Дүние таксондарымен (максимум Pi ≈ 0.04) салыстырғанда пластомның жалпы өзгергіштігін жоғары көрсетті (максимум Pi ≈ 0.06). Шамасындағы осы айырмашылықтарға қарамастан, екі топта да дәл сол локустар (trnH--psbA, atpH--atpI, accD, rpl32--trnL, ycf1) өзгергіштік шыңы ретінде көрінді, бұл аталған аймақтардың географиялық шығу тегіне қарамастан Salvia бойынша тұрақты түрде ақпараттылығын көрсетеді.
DiscussionОбсуждениеТалқылау
Plastome Evolution and Nucleotide Diversity PatternsЭволюция пластома и закономерности нуклеотидного разнообразияПластом эволюциясы және нуклеотидтік әртүрлілік заңдылықтары
Our results demonstrate that nucleotide diversity in the Salvia chloroplast genome is strongly partitioned between coding and non-coding regions, with intergenic spacers exhibiting substantially higher variation than protein-coding sequences. This pattern is consistent with expectations based on functional constraint: coding regions, particularly those encoding photosynthetic and ribosomal proteins, are under strong purifying selection to maintain protein function, whereas intergenic spacers experience relaxed constraint and accumulate substitutions more freely (Kelchner, 2000; Dong et al., 2012).Наши результаты показывают, что нуклеотидное разнообразие в хлоропластном геноме Salvia чётко распределяется между кодирующими и некодирующими областями, причём межгенные спейсеры демонстрируют существенно более высокую изменчивость по сравнению с белок-кодирующими последовательностями. Эта закономерность согласуется с ожиданиями, основанными на функциональных ограничениях: кодирующие области, особенно кодирующие фотосинтетические и рибосомные белки, находятся под сильным очищающим отбором, поддерживающим функцию белка, тогда как межгенные спейсеры испытывают ослабленные ограничения и свободнее накапливают замены (Kelchner, 2000; Dong et al., 2012).Біздің нәтижелеріміз Salvia хлоропласт геномындағы нуклеотидтік әртүрлілік кодтаушы және кодтамайтын аймақтар арасында айқын бөлінетінін, ал гендікаралық спейсерлер белок кодтаушы тізбектермен салыстырғанда едәуір жоғары өзгергіштік көрсететінін айқындайды. Бұл заңдылық функционалдық шектеулерге негізделген болжамдарға сәйкес келеді: кодтаушы аймақтар, әсіресе фотосинтетикалық және рибосомалық белоктарды кодтайтындар, белок қызметін сақтайтын күшті тазартушы сұрыпталу астында болады, ал гендікаралық спейсерлер босаңсыған шектеулерді сезінеді және алмасуларды еркінірек жинақтайды (Kelchner, 2000; Dong et al., 2012).
The elevated diversity observed in the SSC region relative to the LSC and IR regions has been documented in other angiosperm lineages and likely reflects both the absence of stabilizing selection typical of IR-mediated gene conversion and the presence of genes (ndh complex, ycf1) that are less functionally constrained than photosystem components. The gene ycf1, which encodes a large protein involved in protein translocation, has been widely recognized as one of the most variable plastid loci and has proven useful for species-level phylogenetics across multiple plant families, including Lamiaceae (Chen et al., 2023).Повышенное разнообразие, наблюдаемое в области SSC по сравнению с областями LSC и IR, было задокументировано у других линий покрытосеменных растений и, вероятно, отражает как отсутствие стабилизирующего отбора, характерного для IR-опосредованной генной конверсии, так и присутствие генов (комплекс ndh, ycf1), которые менее функционально ограничены, чем компоненты фотосистем. Ген ycf1, кодирующий крупный белок, участвующий в транслокации белков, широко признан одним из наиболее изменчивых пластидных локусов и оказался полезным для филогенетики на видовом уровне у различных семейств растений, включая Lamiaceae (Chen et al., 2023).LSC және IR аймақтарымен салыстырғанда SSC аймағында байқалатын жоғары әртүрлілік басқа жабық тұқымды өсімдік тармақтарында да құжатталған және бұл IR-аралық генетикалық конверсияға тән тұрақтандырушы сұрыпталудың болмауын да, фотожүйе компоненттеріне қарағанда функционалдық тұрғыдан азырақ шектелген гендердің (ndh кешені, ycf1) болуын да көрсетеді. Белоктардың транслокациясына қатысатын ірі белокты кодтайтын ycf1 гені ең өзгергіш пластидтік локустардың бірі ретінде кеңінен танылған және Lamiaceae қоса алғанда, бірнеше өсімдік тұқымдастарында түр деңгейіндегі филогенетика үшін пайдалы болып шықты (Chen et al., 2023).
Evolutionary History and Geographic ContextЭволюционная история и географический контекстЭволюциялық тарих және географиялық контекст
The observed differences in nucleotide diversity between Old World and New World Salvia are best explained by differences in evolutionary age and diversification history. Old World lineages, particularly those from Central Asia and the Mediterranean, represent older clades that have had more time to accumulate plastome variation. Phylogenetic and biogeographic analyses (e.g., Chen et al., 2022; Moein et al., 2023) indicate that Salvia originated in the Old World, likely in the Irano-Turanian region, during the Oligocene to early Miocene. These lineages subsequently underwent extensive diversification across temperate Eurasia and the Mediterranean, providing ample time for mutation accumulation.Наблюдаемые различия в нуклеотидном разнообразии между видами Salvia Старого и Нового Света лучше всего объясняются различиями в эволюционном возрасте и истории диверсификации. Линии Старого Света, особенно происходящие из Центральной Азии и Средиземноморья, представляют собой более древние клады, у которых было больше времени для накопления изменчивости пластома. Филогенетические и биогеографические анализы (например, Chen et al., 2022; Moein et al., 2023) указывают, что род Salvia возник в Старом Свете, вероятно, в Ирано-Туранском регионе, в период от олигоцена до раннего миоцена. Впоследствии эти линии претерпели обширную диверсификацию по умеренной Евразии и Средиземноморью, что обеспечило достаточное время для накопления мутаций.Ескі дүние мен Жаңа дүние Salvia түрлері арасында байқалатын нуклеотидтік әртүрлілік айырмашылықтары эволюциялық жас пен диверсификация тарихындағы айырмашылықтармен жақсы түсіндіріледі. Ескі дүние тармақтары, әсіресе Орталық Азия мен Жерорта теңізі аймағынан шыққандары, пластом өзгергіштігін жинақтауға көбірек уақыты болған көне кладаларды білдіреді. Филогенетикалық және биогеографиялық талдаулар (мысалы, Chen et al., 2022; Moein et al., 2023) Salvia тұқымдасының Ескі дүниеде, ықтимал Иран-Тұран аймағында, олигоценнен ерте миоценге дейінгі кезеңде пайда болғанын көрсетеді. Кейіннен бұл тармақтар қоңыржай Еуразия мен Жерорта теңізі бойынша кең диверсификациядан өтіп, мутацияларды жинақтауға жеткілікті уақыт берді.
In contrast, New World Salvia, particularly the large subgenus Calosphace, represents a much younger radiation. Molecular dating studies (Kriebel et al., 2019) estimate that the major diversification of Calosphace occurred around 15 Ma, with dispersal from Mexico into Central and South America beginning no earlier than ~12 Ma and continuing into the Late Miocene and Pliocene. Consequently, New World lineages have had approximately half the time available for plastome divergence compared to their Old World relatives, which is reflected in the lower maximum Pi values observed in our analyses.Напротив, виды Salvia Нового Света, особенно крупный подрод Calosphace, представляют собой гораздо более молодую радиацию. Исследования молекулярной датировки (Kriebel et al., 2019) оценивают, что основная диверсификация Calosphace произошла около 15 млн лет назад, при этом расселение из Мексики в Центральную и Южную Америку началось не ранее ~12 млн лет назад и продолжалось в позднем миоцене и плиоцене. Вследствие этого у линий Нового Света было примерно вдвое меньше времени для расхождения пластома по сравнению с их родственниками из Старого Света, что отражается в более низких максимальных значениях Pi, наблюдаемых в наших анализах.Керісінше, Жаңа дүние Salvia түрлері, әсіресе ірі Calosphace туысшасы, әлдеқайда жас радиацияны білдіреді. Молекулалық даталау зерттеулері (Kriebel et al., 2019) Calosphace негізгі диверсификациясы шамамен 15 млн жыл бұрын болғанын, ал Мексикадан Орталық және Оңтүстік Америкаға таралу ~12 млн жыл бұрыннан ерте емес басталып, кеш миоцен мен плиоценге дейін жалғасқанын бағалайды. Соның салдарынан Жаңа дүние тармақтарында Ескі дүние туыстарымен салыстырғанда пластом дивергенциясы үшін шамамен екі есе аз уақыт болды, бұл біздің талдауларымызда байқалған төменірек максималды Pi мәндерінде көрініс табады.
Additionally, environmental factors may have influenced the rate of plastome evolution. Central Asian Salvia species often inhabit arid and semi-arid environments characterized by strong climatic seasonality and variable water availability. Such conditions may have promoted adaptive diversification and potentially accelerated the fixation of plastid mutations linked to photosynthetic efficiency or stress tolerance, though direct evidence for adaptive plastome evolution in Salvia remains limited.Кроме того, на скорость эволюции пластома могли повлиять факторы окружающей среды. Центральноазиатские виды Salvia часто обитают в аридных и полуаридных условиях, характеризующихся выраженной климатической сезонностью и переменной доступностью воды. Такие условия могли способствовать адаптивной диверсификации и потенциально ускорять закрепление пластидных мутаций, связанных с эффективностью фотосинтеза или устойчивостью к стрессу, хотя прямые доказательства адаптивной эволюции пластома у Salvia остаются ограниченными.Сонымен қатар, пластом эволюциясының жылдамдығына қоршаған орта факторлары әсер еткен болуы мүмкін. Орталық Азия Salvia түрлері көбінесе айқын климаттық маусымдылықпен және судың өзгермелі қолжетімділігімен сипатталатын құрғақ және жартылай құрғақ ортада мекендейді. Мұндай жағдайлар бейімделгіш диверсификацияға ықпал етіп, фотосинтез тиімділігімен немесе стреске төзімділікпен байланысты пластидтік мутациялардың бекітілуін ықтимал жеделдеткен болуы мүмкін, дегенмен Salvia түрлеріндегі бейімделгіш пластом эволюциясының тікелей дәлелдері әлі де шектеулі.
Implications for Marker Selection and Phylogenetic InferenceЗначение для выбора маркеров и филогенетического анализаМаркер таңдау және филогенетикалық тұжырым үшін маңызы
The identification of high-variability loci provides a clear, data-driven basis for selecting molecular markers tailored to specific phylogenetic or population-genetic questions. For species-level phylogenetics and DNA barcoding within Central Asian Salvia, the intergenic spacers ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, and rpl32--trnL-UAG offer superior resolution compared to traditional single-locus barcodes such as matK or rbcL. These regions not only exhibit high Pi values but also show consistent variability across different Salvia lineages, making them broadly applicable for molecular discrimination.Выявление высокоизменчивых локусов обеспечивает чёткую, основанную на данных базу для выбора молекулярных маркеров, ориентированных на конкретные филогенетические или популяционно-генетические задачи. Для филогенетики на видовом уровне и ДНК-штрихкодирования центральноазиатских Salvia межгенные спейсеры ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2 и rpl32--trnL-UAG обеспечивают более высокое разрешение по сравнению с традиционными однолокусными штрихкодами, такими как matK или rbcL. Эти области не только демонстрируют высокие значения Pi, но и проявляют устойчивую изменчивость у различных линий Salvia, что делает их широко применимыми для молекулярной дискриминации.Жоғары өзгергіш локустарды анықтау нақты филогенетикалық немесе популяциялық-генетикалық мәселелерге бағытталған молекулалық маркерлерді таңдау үшін деректерге негізделген айқын негіз береді. Орталық Азия Salvia түрлерінде түр деңгейіндегі филогенетика мен ДНҚ-штрихкодтау үшін ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2 және rpl32--trnL-UAG гендікаралық спейсерлері matK немесе rbcL сияқты дәстүрлі бір локусты штрихкодтармен салыстырғанда жоғары ажыратымдылық ұсынады. Бұл аймақтар тек жоғары Pi мәндерін көрсетіп қана қоймай, әртүрлі Salvia тармақтарында тұрақты өзгергіштік те көрсетеді, бұл оларды молекулалық ажырату үшін кеңінен қолданылатын етеді.
Among protein-coding genes, ycf1, matK, rpl16, rpl22, and ndhF represent the best candidates for phylogenetic reconstruction at deeper taxonomic levels (e.g., subgenus or section). The gene ycf1, in particular, combines high variability with sufficient length (~5--6 kb) to provide robust phylogenetic signal and has been successfully employed in studies of Lamiaceae phylogeny (Wu et al., 2021; Zhao et al., 2020b).Среди белок-кодирующих генов ycf1, matK, rpl16, rpl22 и ndhF представляют собой наилучших кандидатов для филогенетической реконструкции на более глубоких таксономических уровнях (например, подрода или секции). В частности, ген ycf1 сочетает высокую изменчивость с достаточной длиной (~5--6 kb), что обеспечивает надёжный филогенетический сигнал, и успешно применялся в исследованиях филогении Lamiaceae (Wu et al., 2021; Zhao et al., 2020b).Белок кодтаушы гендердің ішінде ycf1, matK, rpl16, rpl22 және ndhF тереңірек таксономиялық деңгейлерде (мысалы, туысша немесе секция) филогенетикалық реконструкция үшін ең үздік үміткерлер болып табылады. Атап айтқанда, ycf1 гені жоғары өзгергіштікті сенімді филогенетикалық сигнал беретін жеткілікті ұзындықпен (~5--6 kb) ұштастырады және Lamiaceae филогениясын зерттеуде сәтті қолданылған (Wu et al., 2021; Zhao et al., 2020b).
It is important to note that the informativeness of any marker depends on the evolutionary scale of the question. For shallow-level analyses (e.g., intraspecific population structure), the most variable intergenic spacers will be essential. For deeper phylogenetic questions (e.g., relationships among subgenera), moderately variable coding genes such as matK and ndhF may be more suitable, as they accumulate substitutions at a slower rate and are less prone to homoplasy.Важно отметить, что информативность любого маркера зависит от эволюционного масштаба рассматриваемого вопроса. Для анализов на неглубоком уровне (например, внутривидовой популяционной структуры) необходимы наиболее изменчивые межгенные спейсеры. Для более глубоких филогенетических вопросов (например, отношений между подродами) более подходящими могут оказаться умеренно изменчивые кодирующие гены, такие как matK и ndhF, поскольку они накапливают замены с меньшей скоростью и менее подвержены гомоплазии.Кез келген маркердің ақпараттылығы қарастырылатын мәселенің эволюциялық ауқымына байланысты екенін атап өткен маңызды. Таяз деңгейдегі талдаулар үшін (мысалы, түр ішілік популяция құрылымы) ең өзгергіш гендікаралық спейсерлер қажет болады. Тереңірек филогенетикалық мәселелер үшін (мысалы, туысшалар арасындағы байланыстар) matK және ndhF сияқты орташа өзгергіш кодтаушы гендер қолайлырақ болуы мүмкін, өйткені олар алмасуларды баяуырақ жинақтайды және гомоплазияға азырақ бейім.
Future DirectionsПерспективы дальнейших исследованийБолашақ зерттеу бағыттары
While the present study provides a comprehensive plastome-wide diversity profile for Salvia, several avenues for future research remain. First, the development and empirical testing of universal or semi-universal primers targeting the high-variability loci identified here will be essential to translate these findings into practical tools for field-based studies and herbarium-based DNA barcoding initiatives. Second, integration of plastome data with nuclear markers (e.g., low-copy nuclear genes, restriction-site associated DNA sequencing) would provide a more complete picture of Salvia evolutionary history, particularly in cases where plastid introgression or incomplete lineage sorting may confound phylogenetic inference.Хотя настоящее исследование предоставляет всеобъемлющий профиль разнообразия в масштабе всего пластома Salvia, остаётся несколько направлений для дальнейших исследований. Во-первых, разработка и эмпирическая проверка универсальных или полууниверсальных праймеров, нацеленных на выявленные здесь высокоизменчивые локусы, будут необходимы для преобразования этих результатов в практические инструменты для полевых исследований и гербарных инициатив по ДНК-штрихкодированию. Во-вторых, интеграция данных пластома с ядерными маркерами (например, ядерными генами с низким числом копий, секвенированием ДНК, ассоциированным с сайтами рестрикции) обеспечила бы более полную картину эволюционной истории Salvia, особенно в случаях, когда пластидная интрогрессия или неполная сортировка линий могут искажать филогенетический анализ.Бұл зерттеу Salvia түрлерінің бүкіл пластом ауқымындағы әртүрлілігінің жан-жақты профилін ұсынғанымен, болашақ зерттеулер үшін бірнеше бағыт қалып отыр. Біріншіден, осы жерде анықталған жоғары өзгергіш локустарға бағытталған әмбебап немесе жартылай әмбебап праймерлерді әзірлеу және эмпирикалық тексеру осы нәтижелерді далалық зерттеулер мен гербарийге негізделген ДНҚ-штрихкодтау бастамалары үшін практикалық құралдарға айналдыруда маңызды болады. Екіншіден, пластом деректерін ядролық маркерлермен (мысалы, төмен көшірмелі ядролық гендер, рестрикция сайттарымен байланысты ДНҚ секвенирлеу) біріктіру Salvia эволюциялық тарихының толығырақ суретін, әсіресе пластидтік интрогрессия немесе тармақтарды толық емес сұрыптау филогенетикалық тұжырымды бұрмалауы мүмкін жағдайларда, қамтамасыз етер еді.
Third, expanding geographic and taxonomic sampling to include additional Central Asian endemics and rare taxa will improve the representativeness of diversity estimates and may reveal region-specific variability hotspots not detected in the present analysis. Finally, population-level sampling of selected species across environmental gradients would enable investigation of potential adaptive signals in plastome variation, complementing the neutral evolutionary processes that predominantly shape chloroplast genome evolution.В-третьих, расширение географической и таксономической выборки с включением дополнительных центральноазиатских эндемиков и редких таксонов повысит репрезентативность оценок разнообразия и может выявить специфичные для региона «горячие точки» изменчивости, не обнаруженные в настоящем анализе. Наконец, отбор образцов на популяционном уровне у отдельных видов вдоль экологических градиентов позволил бы исследовать потенциальные адаптивные сигналы в изменчивости пластома, дополняя нейтральные эволюционные процессы, которые преимущественно формируют эволюцию хлоропластного генома.Үшіншіден, қосымша Орталық Азия эндемиктері мен сирек таксондарын қамту үшін географиялық және таксономиялық іріктемені кеңейту әртүрлілік бағаларының өкілдігін арттырады және осы талдауда анықталмаған аймаққа тән өзгергіштіктің ыстық нүктелерін аша алады. Соңында, таңдалған түрлерден экологиялық градиенттер бойынша популяция деңгейінде үлгілер алу пластом өзгергіштігіндегі ықтимал бейімделгіш сигналдарды зерттеуге мүмкіндік беріп, хлоропласт геномының эволюциясын негізінен қалыптастыратын бейтарап эволюциялық процестерді толықтырар еді.
ConclusionsВыводыҚорытынды
In summary, our nucleotide-diversity profiling of the Salvia chloroplast genome reveals that variation is concentrated in intergenic spacers and a subset of protein-coding genes, with substantial differences in overall diversity between Old World and New World lineages reflecting their differing evolutionary ages. The high-variability loci identified here—ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, ycf1, matK, rpl16, rpl22, and ndhF—provide a robust, empirically validated foundation for primer development, DNA barcoding, and improved phylogenetic inference in Salvia. These findings support future taxonomic, biogeographic, and conservation studies of Central Asian plant diversity and demonstrate the value of plastome-scale analyses for optimizing marker selection in complex and rapidly evolving plant groups.Таким образом, проведённое нами профилирование нуклеотидного разнообразия хлоропластного генома Salvia показывает, что изменчивость сосредоточена в межгенных спейсерах и части белок-кодирующих генов, причём существенные различия в общем разнообразии между линиями Старого и Нового Света отражают их различный эволюционный возраст. Выявленные здесь высокоизменчивые локусы — ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, ycf1, matK, rpl16, rpl22 и ndhF — обеспечивают надёжную, эмпирически подтверждённую основу для разработки праймеров, ДНК-штрихкодирования и повышения точности филогенетического анализа Salvia. Эти результаты поддерживают будущие таксономические, биогеографические и природоохранные исследования разнообразия растений Центральной Азии и демонстрируют ценность анализов в масштабе пластома для оптимизации отбора маркеров в сложных и быстро эволюционирующих группах растений.Қорытындылай келе, біздің Salvia хлоропласт геномының нуклеотидтік әртүрлілігін профильдеуіміз өзгергіштіктің межгендік спейсерлерде және белок кодтаушы гендердің бір бөлігінде шоғырланғанын көрсетеді, мұнда Ескі және Жаңа Дүние тармақтары арасындағы жалпы әртүрліліктегі айтарлықтай айырмашылықтар олардың әртүрлі эволюциялық жасын көрсетеді. Осы жерде анықталған жоғары өзгергіш локустар — ccsA--ndhD, trnG-GCC--trnfM-CAU, trnW-CCA--trnP-UGG, trnH-GUG--psbA, atpI--rps2, rpl32--trnL-UAG, ycf1, matK, rpl16, rpl22 және ndhF — Salvia түрлерінде праймер әзірлеу, ДНҚ-штрихкодтау және филогенетикалық тұжырымды жақсарту үшін сенімді, эмпирикалық тұрғыдан расталған негіз ұсынады. Бұл нәтижелер Орталық Азия өсімдіктерінің әртүрлілігін болашақта таксономиялық, биогеографиялық және табиғат қорғау тұрғысынан зерттеуді қолдайды және күрделі әрі тез эволюцияланатын өсімдік топтарында маркер таңдауды оңтайландыру үшін пластом ауқымындағы талдаулардың құндылығын көрсетеді.
FundingФинансированиеҚаржыландыру
This research was supported by the State Program “Digital Nature: Development of a digital platform for the flora of Central Uzbekistan” implemented by the Institute of Botany of the Academy of Sciences of the Republic of Uzbekistan (2025–2029); the project “Assessing climate change adaptation in endangered plants of Uzbekistan: A DNA barcoding approach” (AL 9224104464); and the project “Molecular-genetic identification of medicinal plant species in the flora of Uzbekistan and Belarus using DNA markers” (FL-7923051878).Исследование выполнено при поддержке Государственной программы «Цифровая природа: создание цифровой платформы флоры Центрального Узбекистана» Института ботаники Академии наук Республики Узбекистан (2025–2029 гг.); проекта «Оценка адаптации исчезающих растений Узбекистана к изменению климата: подход ДНК-штрихкодирования» (AL 9224104464); и проекта «Молекулярно-генетическая идентификация видов лекарственных растений флоры Узбекистана и Беларуси с помощью ДНК-маркеров» (FL-7923051878).Зерттеу Өзбекстан Республикасы Ғылым академиясы Ботаника институтының «Цифрлық табиғат: Орталық Өзбекстан флорасының цифрлық платформасын құру» мемлекеттік бағдарламасы (2025–2029); «Өзбекстанның жойылып бара жатқан өсімдіктерінің климат өзгерісіне бейімделуін бағалау: ДНҚ-штрихкодтау тәсілі» жобасы (AL 9224104464); және «ДНҚ-маркерлерін қолдана отырып Өзбекстан мен Беларусь флорасындағы дәрілік өсімдік түрлерін молекулалық-генетикалық сәйкестендіру» жобасы (FL-7923051878) аясында қолдау тапты.
Author ContributionsВклад авторовАвторлардың үлесі
All authors contributed to the study conception and design, data analysis and interpretation, and the drafting and critical revision of the manuscript, and approved the final version.Все авторы внесли вклад в концепцию и дизайн исследования, анализ и интерпретацию данных, подготовку и критическую доработку рукописи и одобрили окончательную версию.Барлық авторлар зерттеудің тұжырымдамасы мен дизайнына, деректерді талдау мен түсіндіруге, қолжазбаны әзірлеу мен сын тұрғысынан түзетуге үлес қосты және соңғы нұсқаны мақұлдады.
Data & Code AvailabilityДоступность данных и кодаДеректер мен кодқа қолжетімділік
Chloroplast genome sequences used in this study are publicly available from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/). Alignment files and nucleotide diversity profiles are available upon request from the corresponding author.Последовательности хлоропластных геномов, использованные в данном исследовании, находятся в открытом доступе в базе данных NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/). Файлы выравниваний и профили нуклеотидного разнообразия доступны по запросу у корреспондирующего автора.Бұл зерттеуде пайдаланылған хлоропласт геномдарының тізбектері NCBI GenBank дерекқорында ашық қолжетімді (https://www.ncbi.nlm.nih.gov/genbank/). Туралау файлдары мен нуклеотидтік әртүрлілік профильдері корреспондент автордан сұрау бойынша қолжетімді.
AcknowledgmentsБлагодарностиАлғыс сөздер
This research was supported by the State Program "Digital Nature: Development of a digital platform for the flora of Central Uzbekistan" implemented by the Institute of Botany of the Academy of Sciences of the Republic of Uzbekistan for the period 2025--2029. This research was also supported by the project titled "Assessing climate change adaptation in endangered plants of Uzbekistan: A DNA barcoding approach" (AL 9224104464). Additional support was provided by the project titled "Molecular-genetic identification of medicinal plant species in the flora of Uzbekistan and Belarus using DNA markers" (FL-7923051878).Исследование выполнено при поддержке Государственной программы «Цифровая природа: создание цифровой платформы флоры Центрального Узбекистана», реализуемой Институтом ботаники Академии наук Республики Узбекистан в период 2025--2029. Исследование также поддержано проектом «Оценка адаптации исчезающих растений Узбекистана к изменению климата: подход ДНК-штрихкодирования» (AL 9224104464). Дополнительная поддержка была оказана проектом «Молекулярно-генетическая идентификация видов лекарственных растений флоры Узбекистана и Беларуси с помощью ДНК-маркеров» (FL-7923051878).Зерттеу Өзбекстан Республикасы Ғылым академиясы Ботаника институты 2025--2029 кезеңінде жүзеге асыратын «Цифрлық табиғат: Орталық Өзбекстан флорасының цифрлық платформасын құру» мемлекеттік бағдарламасының қолдауымен орындалды. Зерттеуге сондай-ақ «Өзбекстанның жойылып бара жатқан өсімдіктерінің климат өзгерісіне бейімделуін бағалау: ДНҚ-штрихкодтау тәсілі» жобасы (AL 9224104464) қолдау көрсетті. Қосымша қолдауды «ДНҚ-маркерлерін қолдана отырып Өзбекстан мен Беларусь флорасындағы дәрілік өсімдік түрлерін молекулалық-генетикалық сәйкестендіру» жобасы (FL-7923051878) ұсынды.
EthicsЭтикаЭтика
Not applicable. This study used publicly available sequence data from GenBank.Неприменимо. В исследовании использованы общедоступные данные последовательностей из GenBank.Қолданылмайды. Зерттеуде GenBank дерекқорынан алынған жалпыға қолжетімді тізбек деректері пайдаланылды.
Competing InterestsКонфликт интересовМүдделер қақтығысы
The authors declare no competing interests.Авторы заявляют об отсутствии конфликта интересов.Авторлар мүдделер қақтығысы жоқ екенін мәлімдейді.
ReferencesЛитератураӘдебиеттер
- Chen H., Chen H., Wang B., Liu C. (2023) Conserved chloroplast genome sequences of the genus Clerodendrum Linn. (Lamiaceae) as a super-barcode. PLoS ONE, 18(2), e0277809. DOI: 10.1371/journal.pone.0277809.
- Chen Y. P., Turginov O. T., Turdimatovich T. O., Nuraliev M. S., Lazarević P., Drew B. T., Xiang C.-L. (2022) Phylogeny and biogeography of the northern temperate genus Dracocephalum s.l. (Lamiaceae). Cladistics, 38, 429--451. DOI: 10.1111/cla.12502.
- Dong W., Liu J., Yu J., Wang L., Zhou S. (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE, 7(4), e35071. DOI: 10.1371/journal.pone.0035071.
- Gao C., Wu C., Zhang Q., Zhao X., Wu M., Chen R., Zhao Y., Li Z. (2020) Characterization of chloroplast genomes from two Salvia medicinal plants and gene transfer among their mitochondrial and chloroplast genomes. Frontiers in Genetics, 11, 574962. DOI: 10.3389/fgene.2020.574962.
- Katoh K., Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772--780. DOI: 10.1093/molbev/mst010.
- Kelchner S. A. (2000) The evolution of non-coding chloroplast DNA and its application in plant systematics. Annals of the Missouri Botanical Garden, 87(4), 482--498. DOI: 10.2307/2666142.
- Kriebel R., Drew B. T., Drummond C. P., González-Gallegos J. G., Celep F., Mahdjoub M. M., Rose J. P., Xiang C.-L., Hu G.-X., Walker J. B., Lemmon E. M., Lemmon A. R., Sytsma K. J. (2019) Tracking temporal shifts in area, biomes, and pollinators in the radiation of Salvia (sages) across continents: leveraging anchored hybrid enrichment and targeted sequence data. American Journal of Botany, 106(4), 573--597. DOI: 10.1002/ajb2.1268.
- Larsson A. (2014) AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics, 30(22), 3276--3278. DOI: 10.1093/bioinformatics/btu531.
- Moein F., Jamzad Z., Rahiminejad M., et al. (2023) Towards a global perspective for Salvia L.: phylogeny, diversification and floral evolution. Journal of Evolutionary Biology, 36(3), 589--604. DOI: 10.1111/jeb.14149.
- Nyamgerel N., Baasanmunkh Sh., Munkhtulga D., Tugsbilguun T., Oyuntsetseg B., Xiang Ch.-L., Choi H. J. (2025) Characterization of the complete chloroplast genome of Dracocephalum ruyschiana (Lamiaceae) and its phylogenetic analysis. Korean Journal of Plant Taxonomy, 55(1), 44--51. DOI: 10.11110/kjpt.2025.55.1.44.
- Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J. C., et al. (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution, 34(12), 3299--3302. DOI: 10.1093/molbev/msx248.
- Shang M., Wang J., Dai G., Zheng J., Liao B., Wang J., Duan B. (2023) Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Ajuga and common adulterants. Frontiers in Plant Science, 14, 1251829. DOI: 10.3389/fpls.2023.1251829.
- Tajima F. (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics, 105(2), 437--460. DOI: 10.1093/genetics/105.2.437.
- Wu H., Ma P. F., Li H. T., Hu G.-X., Li D. Z. (2021) Comparative plastomic analysis and insights into the phylogeny of Salvia (Lamiaceae). Plant Diversity, 43(1), 15--26. DOI: 10.1016/j.pld.2020.07.004.
- Yu D., Pei Y., Cui N., Zhao G., Hou M., Chen Y., Chen J., Li X. (2023) Comparative and phylogenetic analysis of complete chloroplast genome sequences of Salvia regarding its worldwide distribution. Scientific Reports, 13, 14268. DOI: 10.1038/s41598-023-41198-y.
- Zhao F., Li B., Drew B. T., Chen Y. P., Wang Q., Yu W. B., Liu E. D., Salmaki Y., Peng H., Xiang C. L. (2020a) Leveraging plastomes for comparative analysis and phylogenomic inference within Scutellarioideae (Lamiaceae). PLoS ONE, 15(5), e0232602. DOI: 10.1371/journal.pone.0232602.
- Zhao F., Drew B. T., Chen Y. P., Hu G. X., Li B., Xiang Ch.-L. (2020b) The chloroplast genome of Salvia: genomic characterization and phylogenetic analysis. International Journal of Plant Sciences, 181(8), 812--830. DOI: 10.1086/710083.
- Zhao Y., Chen Y. P., Drew B. T., Zhao F., Almasi M., Turginov O. T., Xiao J. F., Karimi A. G., Salmaki Y., Yu X. Q., Xiang C. L. (2024) Molecular phylogeny and taxonomy of Phlomoides (Lamiaceae, subfamily Lamioideae) in China: insights from molecular and morphological data. Plant Diversity, 46(4), 462--475. DOI: 10.1016/j.pld.2024.04.011.
Copyright © 2026 Nikitina, Ergashov, Yusupov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.